- Author
Haibao Tang (tanghaibao), Brent Pedersen (brentp)
- License
Typically in comparative genomics, we can identify anchors, chain them into syntenic blocks and interpret these blocks as derived from a common descent. However, when comparing two genomes undergone ancient genome duplications (plant genomes in particular), we have large number of blocks that are not orthologous, but are paralogous. This has forced us sometimes to use ad-hoc rules to screen these blocks. So the question is: given the expected depth (quota) along both x- and y-axis, select a subset of the anchors with maximized total score.
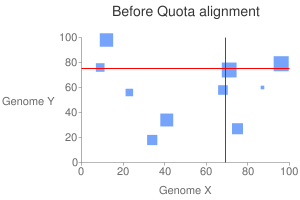
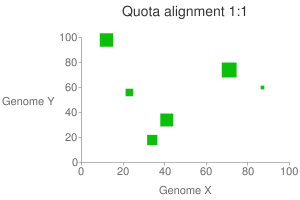
This program tries to screen the clusters based on the depth constraints enforced by the user. For example, between rice-sorghum comparison, we can enforce 1:1 ratio to get all the orthologous blocks; or maybe 4:2 to grab orthologous blocks between athaliana-poplar. But the quota has to be given by the user. The program than tries to optimize the scores of these blocks globally.
To see the algorithm in action without installation, please go to CoGe SynMap tool. Select "Analysis Options", select algorithm options for "Merge Syntenic Blocks" (quota_align.py--merge) and/or "Syntenic Depth" (quota_align.py --quota).
Download the most recent codes at:
git clone http://github.com/tanghaibao/quota-alignment.git
Required dependencies
- Python version >=2.7
- GNU linear programming kit GLPK.
Please put the executable glpsol on the PATH.
Optional dependencies
- SCIP.
Faster integer programming solver, choose the binary (32-bit, 64-bit) that fits your machine. SCIP might have dependency on LAPACK, which needs to be installed too. Please rename the executable scip and put it on the PATH, for example:
sudo cp scip-x.x.0.linux.x86_64 /usr/local/bin/scip
sudo chmod +x !$- bx-python package.
This is only required when user wants to analyze .maf formatted data (use maf_utils.py):
easy_install bx-python- BCBio package.
This is only required when user wants to convert .gff file to .bed format, see section Pre- and post-processing.
Default package comes with the test data for case 1 and 2 in run.sh. More test data set can be downloaded here. Unpack into the folder, and execute run.sh.
First you need to figure out a way to convert the BLAST result into the following format (called .raw format), see section Pre- and post-processing, in particular on filtering BLAST output:
1 6 1 4848 12
1 7 1 4847 10
1 8 1 4847 50
1 9 1 4846 14Where the five columns correspond to chr1, pos1, chr2, pos2, and -log10(E-value). Then we can do something like:
quota_align.py --format=raw --merge --Dm=20 --min_size=5 --quota=2:1 maize_sorghum.qa--merge asks for chaining, distance cutoff --Dm=20 for extending the chain, --min_size=5 for keeping the chains that are long enought; --quota=2:1 turns on the quota-based screening (and asks for two-to-one match, in this case, lineage specific WGD in maize genome, make every 2 maize region matching 1 sorghum region). Note that if you set the quota wrong, e.g. suppose you don't know the quota ratio between maize and sorghum, and you typed 1:1, you will see the coverage reports to be too low:
write (134) clusters to 'data/maize_sorghum.qa.filtered'
genome X coverage: 62.6%
genome Y coverage: 97.5%In this case, genome X (maize) has only slightly over half of the genome aligned, missing the duplicated counterpart.
Most often you will have the .maf file. First convert it to .qa format:
cluster_utils.py --format=maf athaliana_lyrata.maf athaliana_lyrata.qaThen you want to do the chaining and the screening in one step:
quota_align.py --merge --Dm=20000 --quota=1:1 --Nm=40000 athaliana_lyrata.qa--merge asks for chaining, and the distance cutoff --Dm=20000 for extending the chain; --quota=1:1 turns on the quota-based screening (and asks for one-to-one match), and the overlap cutoff --Nm=40000. The reason to specify an overlap cutoff is because the quota-based screening is based on 1D block overlap. Sometimes due to the over-chaining, two blocks will only slightly overlap. Therefore the distance 40000 is how much slight overlap we tolerate.
Finally you can get the screened .maf file by doing:
maf_utils.py athaliana_lyrata.qa athaliana_lyrata.mafYour final screened .maf file is called athaliana_lyrata.maf.filtered. Hint: you can compare the original and filtered .maf using Miller lab's Gmaj tool.
First we need to figure out how to get the input data. See the last two sections for preparing data from BLAST and BLASTZ. Then we can do something like the following:
quota_align.py --format=raw --merge --Dm=20 --min_size=5 --self --quota=2:2 grape_grape.rawThe reason for setting up --quota=2:2 is because grape has paleo-hexaploidy event. Therefore many regions will have 3 copies, but we need to remove the self match. Therefore we should do 2:2 instead. --self option may be turned on for finding paralogous blocks, when you have reduced the redundancies in your .qa file (note that self-match is symmetric across diagonal). The reason for that is in the self-matching case, the constraints on the union of the constraints on both axis, rather than on each axis separately.
For a lineage that has tetraploidy event (genome doubling), using the example of brachypodium (which has undergone an ancient pan-grass tetraploidy), we can do:
quota_align.py --format=raw --merge --Dm=20 --self --quota=1:1 brachy_brachy.rawNote in this case, --quota=1:1 since we have most regions in 2 copies, but we need to ignore the self match. Therefore the rule is when searching paralogous blocks (always do --quota=x:x, where x is the multiplicity minus 1).
This is so far only supported when --quota=1:1. For example:
quota_align.py --merge --quota=1:1 athaliana_lyrata.qa
cluster_utils.py --print_grimm athaliana_lyrata.qa.filteredThe script will print this:
>genome X
1 2 3 4 5 6 7 8 9 10 11$
12 13 14 15 16 17 18 19$
20 21 22 23 24 25 26 27 28 29 30 31$
32 33 34 35 36$
37 38 39 40 41$
42 43 44 45 46 47 48 49 50$
51 52 53 54 55 56 57 58$
59 60 61$
62 63$
>genome Y
-1 2 -3 4 -6 -7 5 8 10 9 11 -14 13 -12 15 16 17 18 -19$
37 38 24 -25 26 29 28 -30 -27 31 32 33 -34 35 36$
-21 -20 22 23 39 40 41$
-50 49 -48 44 46 -45 47 63 -62 -55 -54 53 -52 51$
-42 43 56 57 -58 -59 60 -61$This is the input format for Glenn Tesler's GRIMM software. You can either run it locally or on their website.
There are a few utility scripts included in scripts/ folder.
Most annotation groups only provide .gff file (see gff format) for the annotation of gene models. I often convert the .gff file to a simpler .bed format (see bed format). You can do the following to create the .bed file (BCBio module required):
gff_to_bed.py athaliana.gff >athaliana.bedThis will get protein-coding models and put these in the .bed format. .gff file must be gff3-compatible, otherwise you have to write customized parser (in fact, this is recommended as most .gff file for genome projects are not compatible). .bed format is required for doing BLAST filtering, see below.
The integer programming solver cannot solve large problem instance (say >60000 variables), this mostly will not happen if we filter our anchors carefully (removing redundant and weak anchors). To filter the BLAST results before chaining, using the blast_to_raw.py shipped in this package. Say you have BLAST file (tabular format) ready. You need to do:
blast_to_raw.py athaliana_grape.blastp --qbed=athaliana.bed --sbed=grape.bed --tandem_Nmax=10 --cscore=.5This will convert the BLAST file into the .raw formatted file that quota_align.py can understand (use --format=raw). For your convenience, several BLAST filters are also implemented in blast_to_raw.py. Notice these BLAST filters are optional.
- Remove local dups
Option --tandem_Nmax=10 will group the local dups that are within 10 gene distance. When this option is on, blast_to_raw.py will write new .nolocaldups.bed file, these will substitute your original .bed file from now on.
- Remove repetitive matches
For genes that have many hits, we will adjust the evalue:
adjusted_evalue(A, B) = evalue(A, B) ** ((counts_of_blast / counts_of_genes) / (counts(A) + counts(B)))- Use the cscore filtering
Option --cscore=.5 will keep only the hits that have a good score. See reference for cscore in the supplementary of sea anemone paper. C-score between gene A and B is defined:
cscore(A, B) = score(A, B)/max(best score of A, best score of B)Typically, after the blast_to_raw.py, we can do the quota_align.py directly:
quota_align.py --format=raw --merge --Dm=20 --min_size=5 --quota=4:1 athaliana_grape.rawTo visualize the quota-align.py result, all you need is the .qa.filtered result, and two .bed file (remember if you have removed local dups above, make sure you use the .nolocaldups.bed). As an example:
qa_plot.py --qbed=athaliana.nolocaldups.bed --sbed=grape.nolocaldups.bed athaliana_grape.qa.filteredThis will generate a dot plot that you can stare to spot any problem. Below is an example of athaliana-grape dot plot when quota of 4:1 is enforced (meaning that there are expected 4 athaliana regions mapping to 1 grape region).
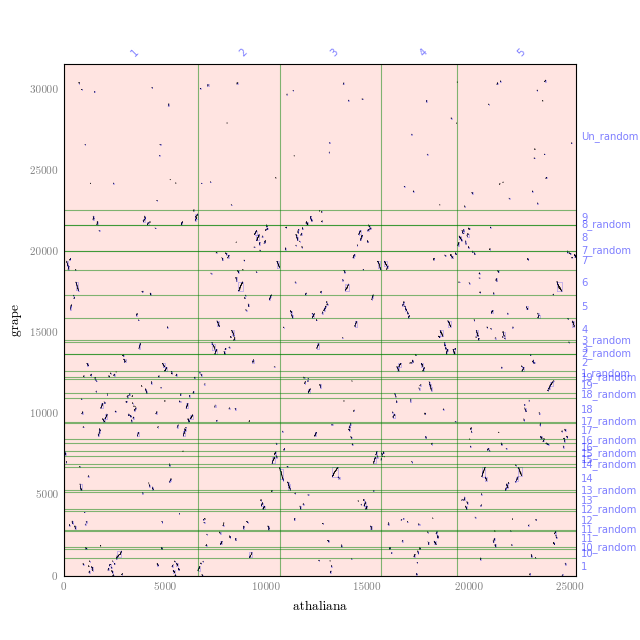
The result of quota-based screening can be compared to the raw blast result. Using the blast_plot.py in script folder. The syntax is similar to qa_plot, only on differernt input format:
blast_plot.py --qbed=athaliana.bed --sbed=grape.bed athaliana_grape.blastpThe following is shell script run.sh that can be used from a BLAST output to the dot plot figure. Please note that you need to modify the path and params to make it work on your machine:
#!/bin/bash
# quota-alignment folder
QA=${HOME}/projects/quota-alignment/
# query species
SA=brapa
### target species
SB=athaliana
# filter blast results (note that it needs to be tab-delimited blast m8 format)
${QA}/scripts/blast_to_raw.py ../blast/${SA}_${SB}.blastz --qbed=${SA}.bed --sbed=${SB}.bed --tandem_Nmax=10 --cscore=0.7
# run the quota-based screening
${QA}/quota_align.py --format=raw --merge --Dm=30 --min_size=5 --quota=3:1 ../blast/${SA}_${SB}.raw
# visualize result as dot plot
${QA}/scripts/qa_plot.py --qbed=${SA}.nolocaldups.bed --sbed=${SB}.nolocaldups.bed ../blast/${SA}_${SB}.raw.filteredTang et al. (2011) Screening synteny blocks in pairwise genome comparisons through integer programming. [ BMC Bioinformatics ]