diff --git a/source/_posts/DeePMD_20_06_2025.md b/source/_posts/DeePMD_20_06_2025.md
new file mode 100644
index 0000000..1c81819
--- /dev/null
+++ b/source/_posts/DeePMD_20_06_2025.md
@@ -0,0 +1,78 @@
+---
+title: "The Era of Large Atom Models | Universal Machine Learning Potential Energy Surfaces for CHON Chemical Reactions"
+
+date: 2025-06-20
+
+categories:
+- DeePMD
+---
+
+Recently, the Beijing Institute for Scientific Intelligence, in collaboration with the Shanghai Institute for Creative Intelligence, the Zhu Tong research group at East China Normal University, and New York University Shanghai, etc., pre-published the latest research progress in the field of large atom models on ChemRxiv under the title "General reactive machine learning potentials for CHON elements".
+
+This study proposes a complete workflow for systematically constructing universal chemical reaction machine learning potential energy surfaces (MLPs) in the era of large atom models. It has breakthroughly built universal reactive MLPs for elements C, H, O, and N. Through innovative data construction and hybrid training strategies, it achieves chemical reaction simulation capabilities approaching DFT accuracy. The team proposed a dynamic sampling method of "wide coverage + active learning", generating the RXN-xTB pre-training dataset composed of over 17 million non-equilibrium structures and the fine-tuning dataset RXN-xTB-AL containing 200,000 structures. Combined with pre-training and Δ-learning collaborative optimization, the hybrid training strategy enables the DPA-3-DF model to achieve an MAE of 0.51 kcal/mol in energy prediction and 0.49 kcal/mol/Å in force prediction, significantly surpassing various existing mainstream neural network architectures. Dynamic simulation verification shows that the model can accurately characterize the dynamic bond fission process of complex reactions, providing a new paradigm that balances quantum accuracy and molecular dynamics efficiency for catalytic design and reaction mechanism analysis. This research achievement marks a major leap in machine learning potential energy in the field of chemical reaction modeling, providing a feasible new path for the precise and efficient simulation of typical organic reactions and catalytic systems.
+
+Paper link:
+https://chemrxiv.org/engage/chemrxiv/article-details/684ffe583ba0887c33dad39b
+
+
+
+## Research Background
+
+In the field of computational chemistry and molecular simulation, accurately describing chemical reaction paths and their potential energy surfaces (PES) has always been a core challenge for understanding reaction mechanisms, designing catalysts, and developing new materials. Traditional quantum chemistry methods such as DFT and CCSD(T) have absolute advantages in accuracy but are difficult to apply to complex reaction networks and large-scale systems due to their high computational costs. In contrast, molecular mechanics and semi-empirical methods are computationally efficient but often fail to cover the process of chemical bond breaking and formation, and cannot accurately describe reactive behavior.
+
+In recent years, machine learning potential energy surfaces (MLPs) have gradually become an important tool connecting high precision and high efficiency by learning complex structure-energy mapping relationships from high-precision quantum chemistry data. Although various MLPs models for equilibrium structures and material systems have made breakthroughs, developing universal reactive MLPs models for chemical reaction systems, especially those with wide coverage, strong generalization, and high precision, still faces huge challenges. Reaction systems involve high-energy transition states, non-equilibrium configurations, and diverse chemical bond rearrangement processes. How to systematically construct high-quality training data and optimize efficient model architectures has become a key scientific issue promoting the development of the field. Focusing on C, H, O, N element systems, this work proposes a systematic data construction, model training, and verification evaluation system, significantly improving the accuracy, robustness, and generalization ability of reactive MLPs.
+
+## How the Model was Developed
+
+**Comprehensive and Efficient Data Construction System:**
+
+**Wide-coverage, multi-scale, dynamic reaction space sampling**
+
+
+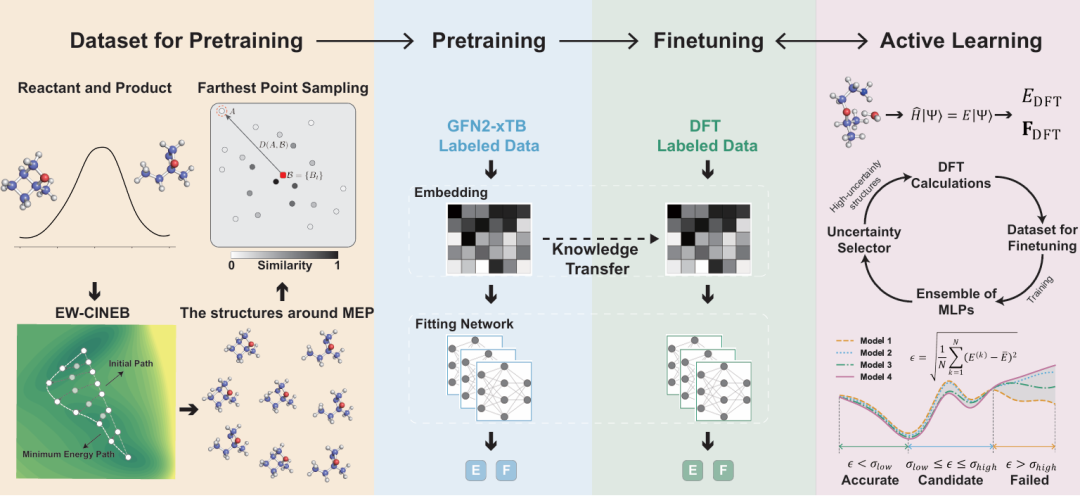 +
+*Figure 1: Model construction process*
+
+
+To effectively capture the diverse non-equilibrium configurations in complex reaction processes, based on the Transition1x and RGD-1 datasets, the authors adopted the NEB reaction path sampling and molecular similarity-guided Farthest Point Sampling (FPS) algorithm to achieve large-scale reaction space coverage, generating the RXN-xTB pre-training dataset composed of over 17 million non-equilibrium structures. Thereafter, through an active learning strategy, high-uncertainty samples were automatically identified, and high-precision DFT labeled data was iteratively supplemented, finally forming the fine-tuning dataset RXN-xTB-AL containing 200,000 structures. This process greatly improves the efficient utilization of data, ensuring wide coverage and balanced distribution of the model in chemical space, laying a solid foundation for subsequent high-precision model training.
+
+## How Powerful is the Model?
+
+**Systematic Training Strategy Optimization:**
+
+**Fully exploiting the collaborative advantages of pre-training and Δ-learning**
+
+
+
+
+
+*Figure 1: Model construction process*
+
+
+To effectively capture the diverse non-equilibrium configurations in complex reaction processes, based on the Transition1x and RGD-1 datasets, the authors adopted the NEB reaction path sampling and molecular similarity-guided Farthest Point Sampling (FPS) algorithm to achieve large-scale reaction space coverage, generating the RXN-xTB pre-training dataset composed of over 17 million non-equilibrium structures. Thereafter, through an active learning strategy, high-uncertainty samples were automatically identified, and high-precision DFT labeled data was iteratively supplemented, finally forming the fine-tuning dataset RXN-xTB-AL containing 200,000 structures. This process greatly improves the efficient utilization of data, ensuring wide coverage and balanced distribution of the model in chemical space, laying a solid foundation for subsequent high-precision model training.
+
+## How Powerful is the Model?
+
+**Systematic Training Strategy Optimization:**
+
+**Fully exploiting the collaborative advantages of pre-training and Δ-learning**
+
+
+
+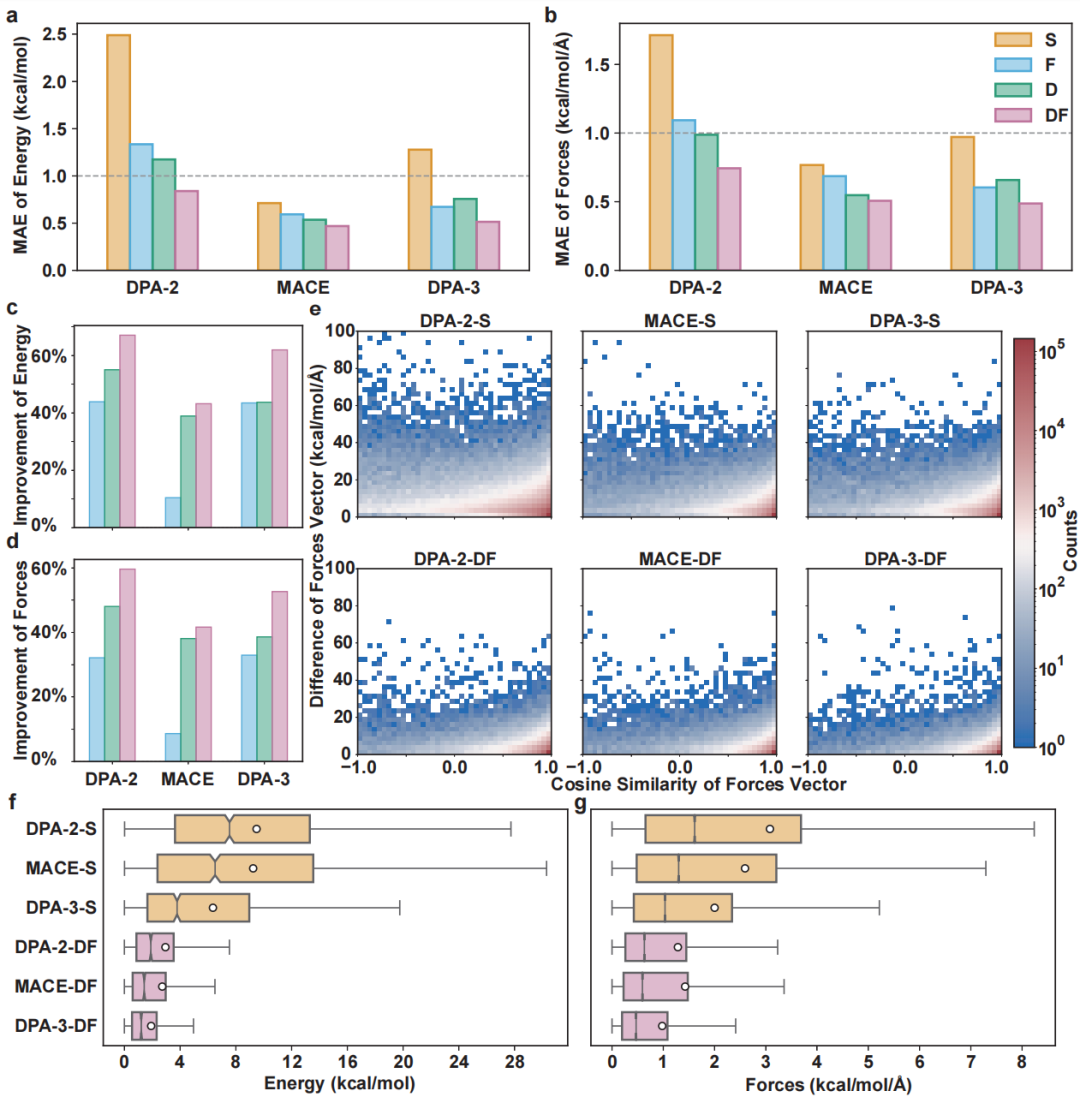 +
+*Figure 2: Model performance comparison under different training strategies*
+
+
+
+In model training, the authors systematically compared the effects of zero-shot training, pre-training fine-tuning, Δ-learning, and their hybrid strategies. The pre-training stage helps the model learn broad molecular representation capabilities, while Δ-learning effectively improves the model's fine-grained characterization of energy and force by learning the differences between semi-empirical low-order methods (GFN2-xTB) and high-order quantum chemistry calculations (DFT). Finally, the hybrid training strategy enables the DPA-3-DF model to achieve an MAE of 0.51 kcal/mol in energy prediction and 0.49 kcal/mol/Å in force prediction, significantly surpassing various existing mainstream neural network architectures. It is worth noting that even in the external distribution test sets of GDB-10 and GDB-17, the model still demonstrates excellent generalization performance, verifying the strong robustness of the overall training strategy.
+
+**System Performance Verification: Energy Barrier and Reaction Energy Prediction**
+
+
+
+
+
+*Figure 2: Model performance comparison under different training strategies*
+
+
+
+In model training, the authors systematically compared the effects of zero-shot training, pre-training fine-tuning, Δ-learning, and their hybrid strategies. The pre-training stage helps the model learn broad molecular representation capabilities, while Δ-learning effectively improves the model's fine-grained characterization of energy and force by learning the differences between semi-empirical low-order methods (GFN2-xTB) and high-order quantum chemistry calculations (DFT). Finally, the hybrid training strategy enables the DPA-3-DF model to achieve an MAE of 0.51 kcal/mol in energy prediction and 0.49 kcal/mol/Å in force prediction, significantly surpassing various existing mainstream neural network architectures. It is worth noting that even in the external distribution test sets of GDB-10 and GDB-17, the model still demonstrates excellent generalization performance, verifying the strong robustness of the overall training strategy.
+
+**System Performance Verification: Energy Barrier and Reaction Energy Prediction**
+
+
+
+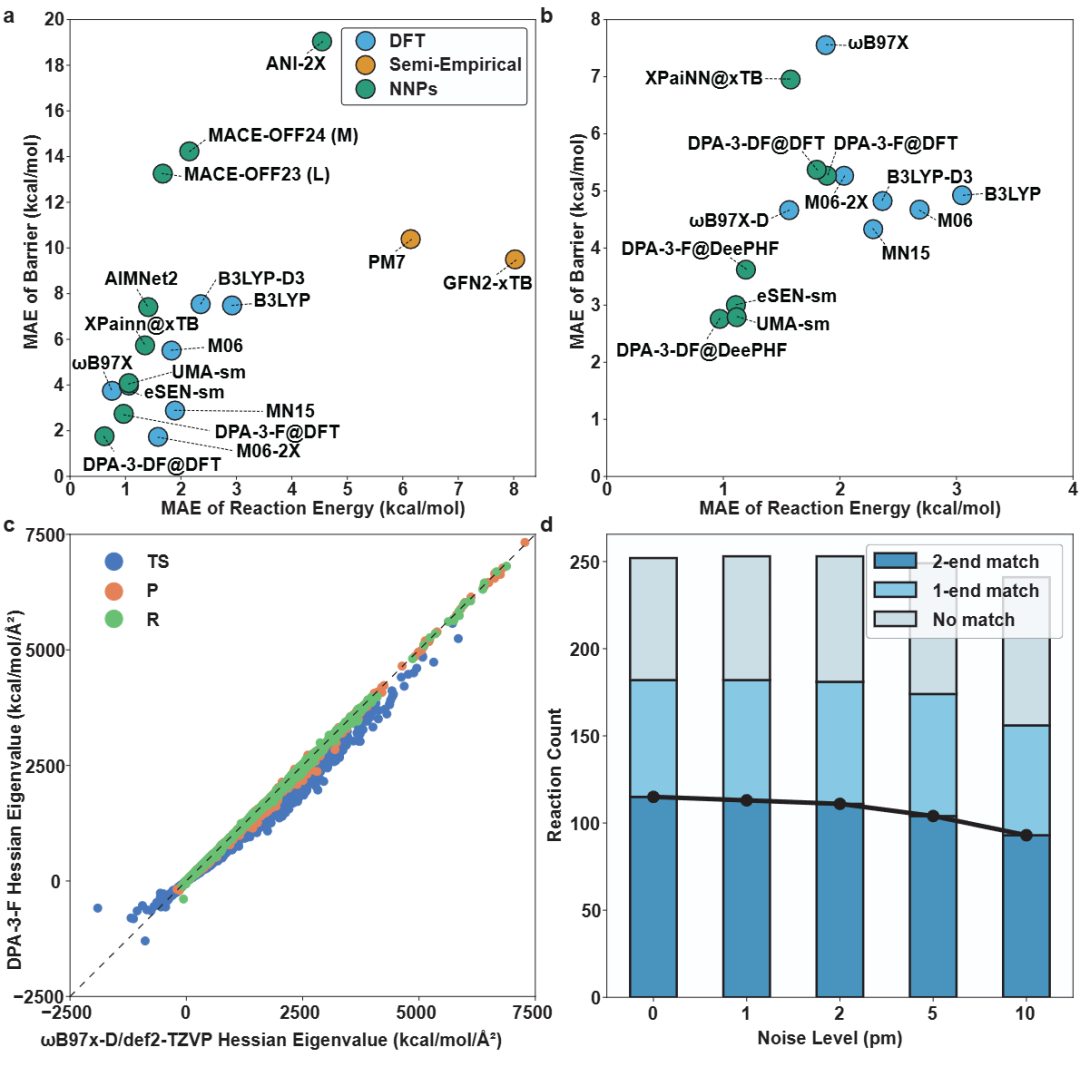 +
+*Figure 3: Systematic performance evaluation*
+
+
+In the actual reaction performance evaluation, the DPA-3-DF model showed leading performance in the GDB-10 reaction test set, with reaction energy prediction errors lower than various DFT methods (M06, B3LYP, etc.), and some barrier prediction indicators have surpassed M06-2X. Further combining the DeePHF model to recalculate data labels and applying a multi-task training framework, the model achieves an MAE of 0.97 kcal/mol in reaction energy prediction, approaching the "chemical accuracy" level. In addition to scalar properties, the authors also evaluated the model's ability to predict the Hessian second derivative matrix. Although the Hessian near the transition state still presents challenges, the model has outperformed some DFT reference methods in stable structures (reactants and products), providing important basic support for subsequent TS search and dynamic modeling.
+
+**Dynamic Reaction Path Simulation**
+
+
+
+
+
+*Figure 3: Systematic performance evaluation*
+
+
+In the actual reaction performance evaluation, the DPA-3-DF model showed leading performance in the GDB-10 reaction test set, with reaction energy prediction errors lower than various DFT methods (M06, B3LYP, etc.), and some barrier prediction indicators have surpassed M06-2X. Further combining the DeePHF model to recalculate data labels and applying a multi-task training framework, the model achieves an MAE of 0.97 kcal/mol in reaction energy prediction, approaching the "chemical accuracy" level. In addition to scalar properties, the authors also evaluated the model's ability to predict the Hessian second derivative matrix. Although the Hessian near the transition state still presents challenges, the model has outperformed some DFT reference methods in stable structures (reactants and products), providing important basic support for subsequent TS search and dynamic modeling.
+
+**Dynamic Reaction Path Simulation**
+
+
+
+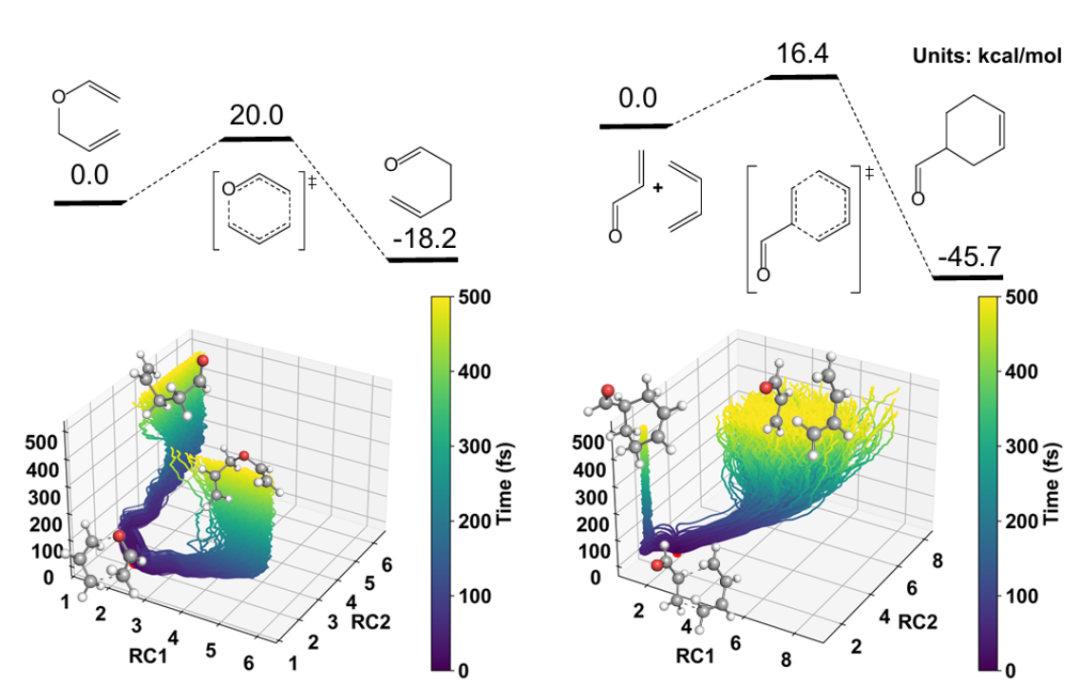 +
+*Figure 4: Verification of reactive molecular dynamics simulation*
+
+
+Finally, to verify the model's application capabilities in real dynamic processes, the authors selected classic Diels-Alder reactions and Claisen rearrangement reactions to carry out molecular dynamics simulations starting from TS. The results show that the model can stably characterize the complex bond breaking and formation behavior in the reaction path, and the trajectories converge to reactant and product states within a limited time scale. This result verifies the model's practical application potential in dynamic reaction network evolution, dynamic process modeling, and complex system simulation, providing solid support for subsequent landing applications in scenarios such as catalysis, materials, and synthetic design.
+
+The full-process development framework proposed in this work forms a closed-loop innovation model in data construction, model training, performance verification, and dynamic application, providing an important paradigm and open resources for the construction of high-precision, strong generalization MLPs models for universal reaction systems. This research achievement is expected to provide powerful tool support for in-depth mechanism research on complex reaction systems, precise catalytic design, and automatic reaction discovery.
+
+
+*Figure 4: Verification of reactive molecular dynamics simulation*
+
+
+Finally, to verify the model's application capabilities in real dynamic processes, the authors selected classic Diels-Alder reactions and Claisen rearrangement reactions to carry out molecular dynamics simulations starting from TS. The results show that the model can stably characterize the complex bond breaking and formation behavior in the reaction path, and the trajectories converge to reactant and product states within a limited time scale. This result verifies the model's practical application potential in dynamic reaction network evolution, dynamic process modeling, and complex system simulation, providing solid support for subsequent landing applications in scenarios such as catalysis, materials, and synthetic design.
+
+The full-process development framework proposed in this work forms a closed-loop innovation model in data construction, model training, performance verification, and dynamic application, providing an important paradigm and open resources for the construction of high-precision, strong generalization MLPs models for universal reaction systems. This research achievement is expected to provide powerful tool support for in-depth mechanism research on complex reaction systems, precise catalytic design, and automatic reaction discovery.
+