
To create a fresh environment for chromatiblock to run in do:
conda create --name chromatiblock
conda activate chromatiblock
conda install chromatiblock --channel conda-forge --channel bioconda
Then in future to run chromatiblock you can reactivate this environment using conda activate chromatiblock
Alternatively you can download and run the script from here.
(these will be installed automatically if chromatiblock is installed with conda)
- Python >= 3.6.0
While chromatiblock will run fine without the following programs, they are needed for certain tasks.
- Sibelia - for automatic generation of colinear blocks
- cairosvg - for creating PNG and PDF images (svg and html supported natively)
- ncbi-blast+ - for automatic annotation of genes
- svg-pan-zoom.min.js - by default the html output uses an online javascript library for zoom functionality, the -pz flag can be used to point to an offline location of svg-pan-zoom.
python chromatiblock -f genome1.fasta genome2.fasta .... genomeN.fasta -w cb_working_dir -o image.svg
Will run chromatiblock on the genomeN.fasta files and create an svg image of the output. Intermediate files will be created
in cb_working_dir.
or
python chromatiblock -d /path/to/fasta_directory/ -w cb_working_dir -o image.html
Will run chromatiblock on all fasta or genbank files in /path/to/fasta_directory/ and create an interactive webpage of the output. Intermediate files will be created
in cb_working_dir.
A tooltip will show block # and location when the mouse is hovered over a block. Other blocks of the same type will also be highlighted. Clicking on a block will show additional information, including the underlying sequence.
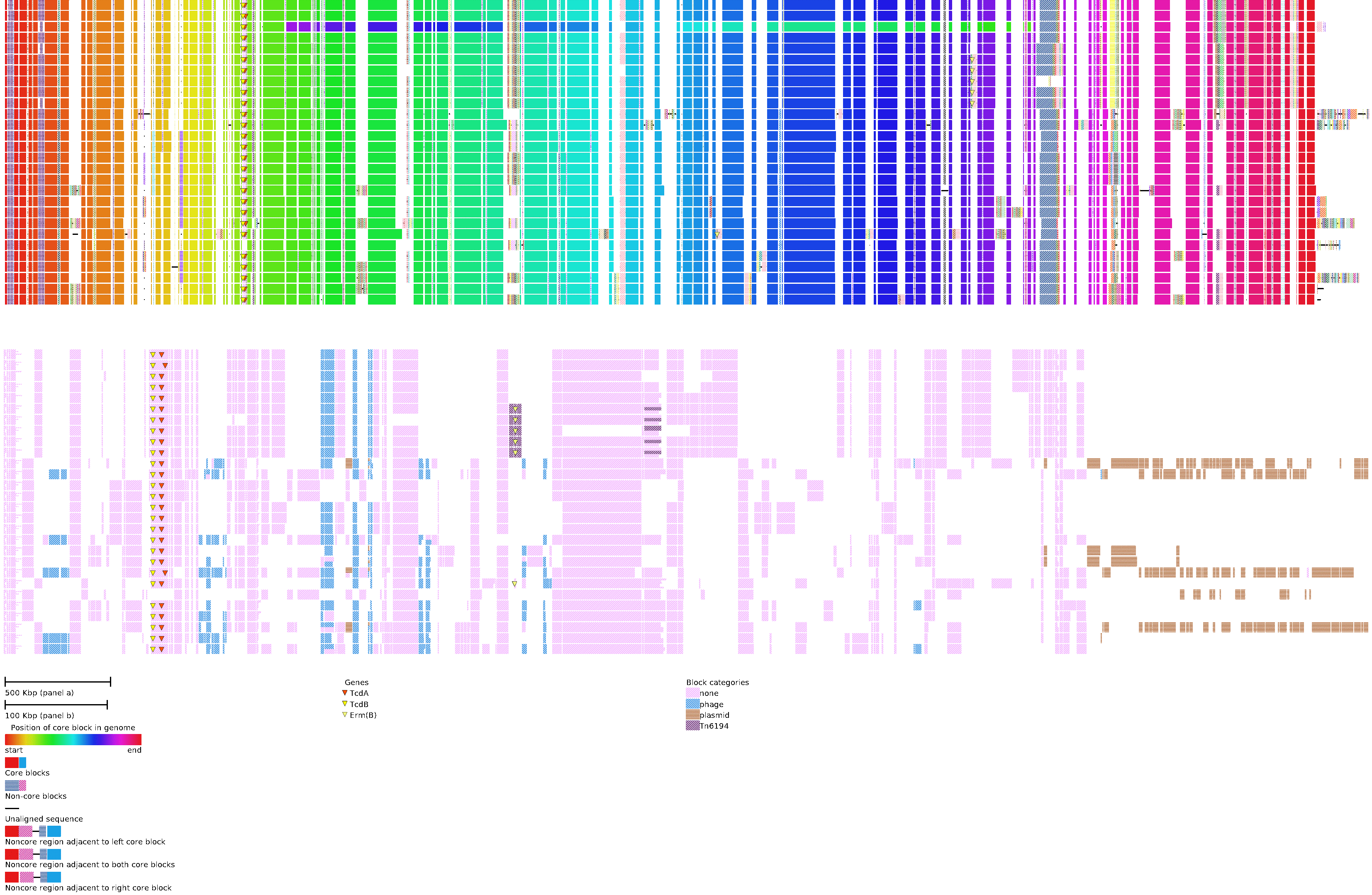
An example of a global alignment of 28 complete C. difficile genomes is shown in the above figure.
TOP) The global alignment view shows the arrangement of core blocks (i.e. syntenic regions found once in all genomes) in the alignment and how non-core blocks (i.e syntenic regions found in 2 or more genomes) and unique sequences (i.e. sequence found in a single genome) are arranged relative to the core blocks. Core blocks are aligned according to their arrangement in the first genome. The color of the core blocks for each genome is determined by its position. Between any two adjacent core blocks there exists a combination of non-core blocks and unique sequence. This combination is grouped and positioned between the two core blocks to which they are adjacent. In instances where the group cannot be placed between its two adjacent core blocks it is placed arbitrarily next to one of the core blocks to which it is adjacent. This is indicated by removing the gap between core and non-core blocks.
In top panel of the above figure a large inversion can be observed in the third isolate from the top, indicated by a difference in ordering of core block colors relative to the reference. Plasmids, found in 9 genomes, consist entirely of non-core and unique blocks. They are positioned on the right side of the figure.
BOTTOM) The alignment difference view shows the presence and absence of all non-core blocks.Each genome is represented as a row and each non-core block is assigned a column in the order they are most commonly found in the genome. Presence of each non-core block is shown as a patterned rectangle in the genomes row. As non-core blocks may be present more than once, duplicates are shown by splitting the blocks according to repeat number. Blocks are coloured by user provided categories.
This example shows that the main C. difficile pathogenicity locus (PaLoc) that contains the genes encoding the TcdA enterotoxin and TcdB cytotoxin, has been lost in the sixth isolate from the bottom. Six isolates also contain a transposon carry-ing the erm(B) gene, encoding a 23S rRNA methyltransferase that confers resistance to erythromycin. The erm(B)gene is also present in the seventh isolate from the bottom, but located on a novel transposon and inserted elsewhere in the genome.
This simple example uses 12 randomly selected complete H. pylori genome to demonstrate how to easily generate a chromatiblock webpage.
Files to generate the H. pylori example can be found here (n.b. this dataset is a lot smaller and will run quicker).
The expected output of this example is available here.
To run: extract files to your current directory and then use the command
chromatiblock -d simple_demo -w cb_working_dir_simple -o H_pylori.html -l simple_demo/order_list.txt
-d - directory to look for fasta files
-w - working directory location
-o - output file, for an svg use .svg suffix
-l - How to order fasta files in figure
Then open H_pylori.html with your favourite browser.
This demo illustrates how to add gene symbols and colour blocks by categories. It uses 28 genomes generated as part of the pathogen surveillance program at Mount Sinai Hospital.
Files to generate the example figure can be found here.
To run: extract files to your current directory and then use the command
chromatiblock -d advanced_demo -w cb_working_dir_advanced -o C_difficile.html -l advanced_demo/order_list.txt -gb advanced_demo/toxins.faa -c advanced_demo/categories.tsv
-d - directory to look for fasta files
-w - working directory location
-o - output file, for an svg use .svg suffix
-l - How to order fasta files in figure
-gb - Fasta file of proteins to annotate in genome, genes will be found using BLASTx
-c - custom colours to use for bottom panel of figure
-h, --help
show this help message and exit
-d, --input_directory </path/to/fastas/>
Directory of fasta or genbank files to use as input (will ignore files without .fasta, .fa, .fna, .gb or .gbk suffixes).
-l, --order_list <list_of_filenames.txt>
file containing list of filenames (one per line) in desired order
-f, --fasta_files <genome_1.fasta> <genome_2.fasta> .... <genome_x.fasta>
List of fasta/genbank files to use as input
-w, --working_directory <working_dir>
Folder to write intermediate files.
-s, --sibelia_path </path/to/sibelia/>
Specify path to sibelia (does not need to be set if Sibelia binary is in path)
-sm, --sibelia_mode <loose>
mode for running sibelia <loose|fine|far>
default: loose
-o, --out <outfile.svg>
Location to write output (options *.svg/*.html/*.png/*.pdf) will default to svg (and add extension). (n.b. PDF does not work particularly well all the time)
-q, --ppi <integer>
pixels per inch (only used for png, figure width is 8 inches)
default: 50
-m, --min_block_size <integer>
Minimum size of syntenic block to display.
default: 1000bp
-c, --categorise <categories.txt>
color blocks by category
provide a file where each line contains
<genome.fasta> <fasta_header> <category_name> <start> <stop>
e.g.
genome_1.fasta contig_1 phage 50000 100000
genome_2.fasta contig_1 phage 80000 120000
genome_2.fasta contig_2 plasmid
genome_3.fasta contig_2 plasmid
genome_3.fasta contig_1 transposon 100000 105000
non-core blocks in panel B will be colored according to category if at least 50% falls within a specified range. Start and stop columns are optional.
-gb, --genes_of_interest_blast <genes.faa>
Uses BLASTx to find genes in chromosome and then marks them in panel a and b with a triangle
-gf, --genes_of_interest_file <genes.txt>
mark genes of interest using a list
provide a file where each line contains
<genome.fasta> <fasta_header> <gene_name> <start>
e.g.
genome_1.fasta contig_1 mrcA 50000
genome_2.fasta contig_1 mrcA 80000
genome_2.fasta contig_2 hlb 20000
-gh, --genome_height <int>
Height of genome blocks in pixels
default: 280
-vg, --gap <int>
gap between genomes
default: 20
-ss, --skip_sibelia
Use sibelia output already in working directory
-sb, --skip_blast
use existing BLASTx file for annotation (must use with -gb flag)
-maf, --maf_alignment <alignment.maf>
use a maf file for multiple alignment instead of Sibelia. MAF files can be produced by SibeliaZ and Mugsy.
-pz, --svg_pan_zoom_location
Location of the svg-pan-zoom javascript library, this can be set to an offline location, or a directory relative to the html.
default: http://ariutta.github.io/svg-pan-zoom/dist/svg-pan-zoom.min.js
-hs, --hue_start
Hue of core blocks at the start of the genome.
-he, --hue_end
Hue of core blocks at the end of the genome.
-v, --version
print version and exit.
-e, --extension
When -d is used for input files, chromatiblock will check against this comma seperated list to determine whether to add file to the list of input sequences.
defaul: fasta,fa,fna,gbk,gb
-of, --output_format
file format to write to, if all is selected --out will be a prefix and extension will be added
valid formats are html, svg, png, pdf, all
all will create a svg, png and pdf
-t, --add_labels
Add the fasta names as labels.
--force
overwrite working directory and output
--keep
keep working directory, desciption of intermediate files created by Sibelia can be found here https://github.com/bioinf/Sibelia/blob/master/SIBELIA.md