More information - How to calculate meanFragmentLength #1
Comments
|
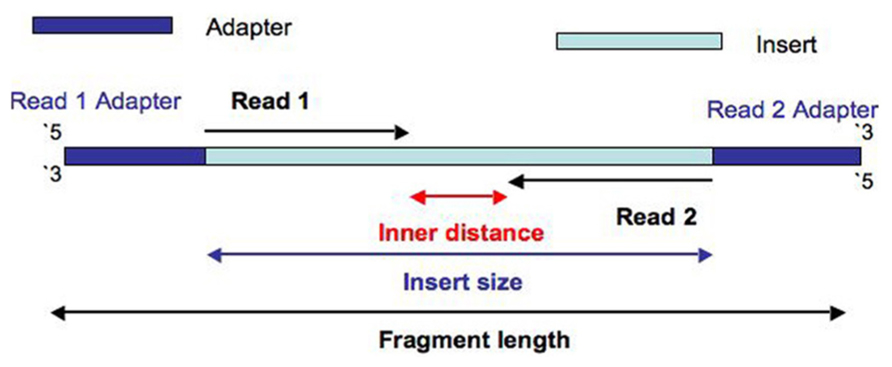
Hi there, I have added an example RNA-seq dataset to the package that can be used to validate your work. See The fragment length is determined by your sequencing protocol. It’s more or less the distance between the two paired reads (including the read lengths, minus any overlap). For a typical long RNA-seq library prep it’s often in the order of 300–600 bp. You can run Picard’s CollectInsertSizeMetrics tool on your aligned BAM files to find the mean fragment length. |
|
Thank you! This is helpful!! |
|
Hi AAlhendi1707, I have searched this problem over and over, and I keep getting routed back to this page. How does one convert from MEAN_INSERT_SIZE to meanFragmentLength? I have seen your posted example, but I am still having trouble understanding this. Any help would be appreciated. Thank you. |
so,that means mean INSERT_SIZE plus 2*adapter? |
|
Can you please explain in detail how to get fragments size from Piccard. If it it insert size + adapter length, Where I will get information related rot adapter length? |
|
The [CollectInsertSizeMetrics(Picard)] tool requires input BAM/SAM file for finding out the mean fragment length. Where as I have only the count matrix file. Is there any way to find the mean fragment length if I only have the count matrix file. |
|
https://www.biostars.org/p/83901/ |
|
I do not think we need to consider insertion size, etc. It evens out in large number. |
|
Hi, I don't understand the output of Picard. How do I get meanFragmentLengthfrom MEAN_INSERT_SIZE? in my case it's a number and it does not make any sense. |
Hello,
Thank you for providing this as a package and will help to streamline the calculations across users. I have seen some students performing wrong calculation.
Can you please include the example data files (counts, annotations, sample_metric)?
Picard CollectInsertSizeMetricshas columns:For the
meanFragmentLengthare you referring to MEAN_INSERT_SIZE column? Just need to confirm since there could be different interpretations for fragment/insert.Thank you.
The text was updated successfully, but these errors were encountered: