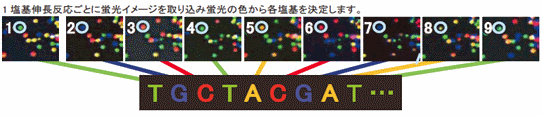
情報・システム研究機構(ROIS)
ライフサイエンス統合データベースセンター(DBCLS)
仲里 猛留 nakazato@dbcls.rois.ac.jp
twitter: @chalkless
2015年5月20日 AJACS御茶ノ水@東京医科歯科大学
AJACS御茶ノ水 > 「次世代シーケンサー(NGS)と関連するデータベース・ツール」
- 次世代シーケンシングデータのコマンドラインによる解析は今日は演習はやりません。
- ∵ 時間がない。マシンパワーとディスク容量が必要なため。データの転送に膨大な時間がかかるため。目的別に解析の方法が違う
- ウェブツールを用いた解析については、この後のコマでとりあげてくれますので、そちらを。
- ↓ 参考サイト
- (Rで)塩基配列解析(主に次世代シーケンサーのデータ) (門田幸二さん:東京大学 大学院農学生命科学研究科 アグリバイオインフォマティクス教育研究ユニット)
- お家でできるMac Bookでやる次世代シーケンスデータ解析 (PDFへのリンク)(緒方法親さん:東京農工大学 応用昆虫学研究室(当時))
- 統合TV:R+Bioconductorを使ったNGS解析1(二階堂愛さん:理化学研究所 発生・再生科学総合研究センター(当時))
- 統合TV:R+Bioconductorを使ったNGS解析2(二階堂愛さん:理化学研究所 発生・再生科学総合研究センター(当時))
- 統合TV:Avadis NGS の使い方~導入編~ … 商用ソフト
- 統合TV:Avadis NGS の使い方~RNA-seq編~ … 商用ソフト
- 統合TV:CLC Workbench シリーズの使い方~導入方法編~ … 別の商用ソフト
- (参考)統合TV:[CLC Workbench シリーズの使い方 ~基本操作編~ … 別の商用ソフト
- 統合TV:CLC Genomics Workbench でショートリードのマッピングを行う … 別の商用ソフト
- バイオインフォマティクス人材育成カリキュラム(次世代シークエンサ)速習コース (NBDC+東大アグリバイオ)… 2週間カンヅメにして、Linuxの使い方から実際の解析まで。統合TVでも収載
- 言葉
- 次世代シーケンサ
- 次世代シーケンサー
- 新型シーケンサ
- New-generation Sequencing (NGS)
- Next-generation Sequiencing (NGS)
- 他にmassively parallel DNA sequencing とか...
- 最近は、 High-throughput DNA sequencing (technology) をよく使う印象(略語はNGS)
- 90年代
- ゲル板
- ポリアクリルアミドゲル電気泳動 + 蛍光標識ダイデオキシヌクレオチド

-
00年代
- キャピラリ
ABI PRISM® 3100-Avant Genetic Analyzerより
- キャピラリ
-
10年代
- NGSの登場
- Sanger法(dideoxy法)→ パイロシーケンシング
- (参考)[原理の動画 (Illumina)](http://www.youtube.com/watch?v=l99aKKHcxC4)
次世代シーケンス解析サービス:概要・原理 | 北海道システム・サイエンス株式会社より
- ようするに顕微鏡+インターバル撮影/タイムラプス撮影 -インターバル撮影/タイムラプス撮影: http://www.youtube.com/watch?v=1Az1YX3GgDw
- 超並列
- どんなの?
- Illumina HiSeq
- Illumina MiSeq
- Ion Torrent
- PacBio
- Togo picture gallery ( http://g86.dbcls.jp/~togoriv/ ) より
© 2011 DBCLS Licensed under CC 表示 2.1 日本 ←クレジットをいれれば、転載・改変・再利用 OK
- Illumina HiSeq




- 【実習】どのくらいのデータ量になるか考えてみましょう
- ゲル板:750 (base/lane) × 48/4 lanes = 9kbase
- キャピラリ:500 (base/lane) × 96 lane = 48kbase
- 次世代: 36 (base/seq) × 300M seq/run = 10.8Gbase = 10,800,000kbase
- ↑これらの数字は規模感をつかむだけなので、ざっくりな数字になっています(1 runにかかる時間は比較してないですし)
- ↑これらの数字は「塩基数」であって、シーケンサの出力である「画像データ」のデータサイズでないことに注意!
- そして、その画像データはSRAには登録されていない
- [参考] 各シーケンサの性能比較
-http://bit.ly/ngsspecbydritoshi (二階堂さん@理研)
- http://dbcls.rois.ac.jp/~kawano/ng.html (河野さん@DBCLS)
- ゲノム、発現解析、メタゲノム解析、ChIP-Seq(転写因子解析)、SNP解析、...
- 目的によって必要なデータ量も違う
- 機器に合った利用を
- 目的によって必要なデータ量も違う


- NGSのデータのレポジトリサイトです
- SRA = Sequence Read Archive
- 昔は「Short Read Archive」だったが、shortでなくなってきたので
- 誰(どこ)が集めているのか?
- NCBI To Discontinue Sequence Read Archive and Peptidome
- 予算がなくなったのでやめます
- 解析結果は受け付けます
- RNA-Seq, ChIP-Seq, and epigenomic data that are submitted to GEO
- Genomic and Transcriptomic assemblies that are submitted to GenBank
- 16S ribosomal RNA data associated with metagenomics that are submitted to GenBank
- EBI、DDBJは直後に続けます宣言
- NCBI SRA(一応)続けられます宣言(11/5/9)
###【実習】DRASearchを使ってみる( http://trace.ddbj.nig.ac.jp/DRASearch/ )
- こういうときはNCBIと思いがちですが、データ転送量が多い + インターフェースきれい なのでDDBJを使いましょう
- http://trace.ddbj.nig.ac.jp/DRASearch/ にアクセス
- Keyword に興味のある語を入れてみましょう(例:variation)
- Filtered by の document type で絞り込み:Study
- Filtered by の organism で絞り込み:Homo sapiens
- ACCESSION の SRP...... をクリック → 詳細が
- 画面右の Navigation にあるFASTQやSRALiteからデータがダウンロード可能
- DDBJにあるドキュメント見てみる
- データ構造(StudyとかExpとかRunとか)
- ↑ これは昔のバージョン。今回は、簡単に理解するためにあえてこれで。 DDBJ Sequence Read Archive - Document - Metadataより
- データ構造(StudyとかExpとかRunとか)
- 実データ:FASTQ形式

@DRR001107.1 GEZQ5FO01EEA7F length=77 GCAACATTCAACACATATGTGTTGAATGTTGCACGACGGNGTGTCGCGTCTCTCAAGGCACACAGGGAGTAGNGNNN +DRR001107.1 GEZQ5FO01EEA7F length=77 C@BBBECCECDBBBAAAAA<441111<?@>?=?????44!00044322====22--..//6998222<7<3/!/!!!
- 1行目: @ + タイトル
- 2行目:塩基配列
- 3行目: + (+ タイトル)
- 4行目:シーケンスクオリティ
- 実データ その2:SRA形式
- FASTQ形式データはディスク容量を食うので圧縮した形式 → SRA ToolkitでFASTQ形式に変換して用いる
- たまに変換にミスることがある。ダウンロードに失敗していないならば、ファイルが壊れてアップされていることがあるので、NCBI(か、手近にDDBJ)にクレームを入れましょう。
統計情報から検索する (DBCLS SRA: http://sra.dbcls.jp/ )
- 統計情報の詳細: http://sra.dbcls.jp/trends.html
- 目的別
- 【実習】興味のある「目的」をクリックしてどんなプロジェクトがあるか見てみましょう
- Platform別
- 【実習】興味のある「Platform」をクリックして、(以下同)
- 生物種別
- 【実習】興味のある「生物種」をクリックして、(以下同)
- 組合わせで
- 【実習】Search SRA data 欄でいくつかの条件を組み合わせて検索してみる
- 目的別
- 個々のプロジェクトの中身を見てみましょう
- Sequencing Profileの欄から各々のRun(例:ERR030856)についてクリックして、詳細を見てみよう。→ FastQCによりQuality Checkをかけた結果です
- 質のいいデータで解析したい → ひとつの基準として論文が出ていれば質は高かろう
- DBCLS SRA の文献リスト: http://sra.dbcls.jp/cgi-bin/publication.cgi
- NGS関連文献とそこで言及されているNGSデータのリスト
- 目的/Platform/生物種で絞り込み可能
- 目的:「使える」データをさがす
- 文献として成果が出ているSRAデータセットをさがしてデータの内容とともに俯瞰する
- 生物種、目的に制限あり
- 文献が出ているもののうち、疾患に関連するものを疾患名でまとめた → 論文が出ていないものについても拡張予定
- DBCLS SRAにアクセス
- 下の方の Search by diseasesから

#ref(NGSflow.png);
- 主にFastQCといったソフトが使われます http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
fastqc -o SRR067385.qc -f fastq SRR067385.fastq - DBCLS SRAではあらかじめFastQCをかけた結果を表示できるように随時、処理をしています(自分でやらなくてよい!)
- その1:既知のゲノムに貼る (Reference Genome Mapping)
- QCの結果をもとに、使わない部分(使い物にならない部分)を除去する ... FASTX-Toolkit などを用いる
- マッピング... tophat2、bowtie、bwa などを用いる(どれを使えばいいか? → 速度と精度、目的によって使い分けます)
bowtie2 -x hg19. -U SRR1294107.fastq > SRR1294107.sam← SRR1294107 を hg19 に対してマッピングしてSAM形式で出力- FASTQ形式 → SAM形式
- マッピング形式の可視化
- 例:Integrative Genomics Viewer IGVを使い倒す ~マッピングデータを可視化する~ http://togotv.dbcls.jp/20140716.html#p01
- SAM形式は必要に応じてバイナリ形式のBAM形式に変換して用います(逆にBAM形式を人間が読めるようにSAM形式にすることもある)
samtools view -Sb SRR1294107.sam -o SRR1294107.bam(SAMからBAMへの変換)
- 発現量の定量 ... cufflinks などを用いる
- control に対してサンプル(KOした、疾患状態、...)の発現はどう違うか? ... cufflinks のうちcuffdiff
cuffdiff ensembl_gene.gtf -o result SRR1294107.bam control.bam←controlに対してSRR1294107での発現がどう違うか?- ここでensembl_gene.gtf にはトランスクリプトームの情報が含まれています(アノテーション情報)
- 転写単位ごとの発現量情報推定 ... cummeRbund (cufflinks の結果を解析するRパッケージ)
- 多分、ここまで行き着かないだろうので、二階堂さんがAJACSで話した内容の統合TVを参考にしてください: http://togotv.dbcls.jp/20120926.html#p01
- その2:一からつなげる(De novo Assemble)
このあとのコマでやります。
- その1で書いたマッピングするところまでは発現解析と同じです(FASTQ → SAM → BAM)
- binding site予測 ... macs2 などを用いる
samtools sort oct4.bam oct4.sort← 下準備:BAMファイルのソートsamtools index oct4.sort.bam← 下準備:インデックス作成macs2 -t Oct4.bowtie.sort.rmRepeat.bam -c GFP.bowtie.sort.rmRepeat.bam -f BAM -g mm -n Oct4 -B -q 0.01 - 高次解析に進みます
- 今回は時間がないと思うので、二階堂さんがAJACSで話した内容の統合TVを参考にしてください: http://togotv.dbcls.jp/20120926.html#p01
- SNV: single nucleotide variation
- その1で書いたマッピングするところまでは基本的に発現解析と同じです(FASTQ → SAM → BAM)
- samtools を用いる場合
- bwaでマッピング、samtoolsでSNP callという流れは割とみながやっている手法です
samtools mpileup -Bugf in_genome.fasta in_sorted.bam | ./bcftools view -bvcg - > out_raw.vcf - 参考:http://ameblo.jp/drosk/entry-11222753481.html
- bwaでマッピング、samtoolsでSNP callという流れは割とみながやっている手法です
- GATKを用いる場合
- GATKはSNP解析、変異解析に特化したソフト
- 流れ
- Indel付近をリマッピング(一塩基のずれが結果に響くので丁寧にマッピングする)
- SNV検出
- アノテーション情報付与 ... SNPEff などを用いる
- SAM/BAM形式 → vcf形式
- 流れ
- FASTQを用意
- DDBJの登録サイト(D-way)で実験条件を記入 → メタデータが作成される
- NGSデータのアップデート
- 一定期間公開を保留することもできます
- 登録サイト D-way
- 登録マニュアル by DDBJ
- チュートリアルムービー
AJACS御茶ノ水 > 「次世代シーケンサー(NGS)と関連するデータベース・ツール」