This readme was created by Jiří Vorel (vorel@cesnet.cz).
Jiří Vorel and Roman Leontovyč lectured the course.
Information given in this course is current as of 30th November 2023.

- Introduction
- Log in to the frontend server
- Basic orientation in your home directory
- System of software modules
- Raw reads and quality control
- Data manipulation
- Genome assembly
This tutorial, in the brief form of a hands-on course, shows how to process and analyse sequencing data using MetaCentrum NGI (National Grid Infrastructure). Participants will be introduced to the basic usage of MetaCentrum, e.g. how to log in to the frontend server, how to manipulate data properly, and how to start an interactive or batch job.
In the practical part of the course, we will use publicly available sequencing data (produced by Illumina and Oxford Nanopore platforms) for the de novo hybrid assembly of the bacterial genome - specifically, Escherichia coli strain A0 34/86 (as described in this paper). Unfortunatelly, processing raw reads, genome assembly and following gene prediction and annotation are processes (especially in the case of larger eukaryotic genomes) that often require time-consuming tuning for optimal parameters and considerable hardware resources.
Important
This course does not aim to create a perfect genome assembly.
Due to the time limitation of this course and its primary focus (how to use grid infrastructure as effectively as possible), we will create a very rough genome draft using a few necessary steps.
The proposal of a comprehensive approach for a "perfect" bacterial genome assembly can be seen here.
Note
Throughout this tutorial, important points are supplemented with links to documentation with more detailed descriptions and explanations.
To get the full potential of this course, each of the participants should be (or should have):
- be a registered user of MetaCentrum (due to the process involving application approval and propagation of a new account, it is necessary to apply for an account no later than a day before the course).
- know login information - username and password (created during the application process).
- have a laptop with a working internet connection.
- be able to log in to the remote server (via SSH protocol).
Note
This course is designed for participants without command-line (CLI) knowledge. But this knowledge is recommended. All commands and shell scripts used during the course are pasted below and can be directly copied.
Note
No data and software tools need to be downloaded or installed before the course. Data will be downloaded during the course, and all software tools (freely available for non-commercial usage) are already available for MetaCentrum users.
As is typical for grid computing, all submitted jobs are sorted into specific queues (mainly based on the amount of requested resources). The combination of the required resources and the current infrastructure load determines the delay between the job submission and the start of the calculation. Very demanding jobs can wait in the queue for several days before all the required resources are free. We will use a special queue MetaSeminar reserved for this course to avoid this delay. This queue employs two ida machines (ida7 and ida25), each with 20 CPU cores and 128 GB RAM.
Important
Each job submitted during this course needs to target this dedicated queue. As you will see later, interactive jobs will include a parameter -q MetaSeminar and batch jobs will include a line #PBS -q MetaSeminar. In both cases, the job scheduler PBSPro will send jobs to this specified queue.
The following data and software tools will be used during the course:
- Illumina paired-end reads (NCBI SRA accession number: SRX20115911).
- Oxford Nanopore reads (NCBI SRA accession number: SRX20115912).
- NCBI SRA Toolkit for downloading sequencing data.
- FastQC for quality control of Illumina reads.
- NanoPlot for quality control of Oxford Nanopore reads.
- Unicycler for genome assembly.
- QUAST for assembly quality assessment.
Note
Access to MetaCentrum is granted free of charge only to members (employees and students) of academic/research institutions of the Czech Republic.
- Documentation
- Monitoring page
- User support contact
- MetaCentrum is an activity of the CESNET association, part of the e-INFRA CZ - research and development e-infrastructure.
Like most computing/data centres, MetaCentrum nodes run exclusively on Linux (mainly Debian) and are controlled via the command line. Linux is preferred for its stability, security, speed, adaptability, and compatibility. Additionally, software tools for life sciences are primarily designed and optimised for Linux.
We will use one of the login servers known as frontend for logging in. Frontend servers are accessible via SSH protocol and serve as a main gateway for the entire infrastructure.
Warning
Frontend servers are virtual machines with limited computational power and primarily serve for basic data inspection and manipulation, preparation of the shell scripts for batch jobs, short compilations, etc. Please do not use them for long and/or demanding calculations (rather, use an interactive job).
Note
MetaCentrum can be accessed worldwide. We do not apply any geoblocking.
The following diagram shows the frontend servers' position (labelled as Login nodes) in the context of other parts of the grid infrastructure.
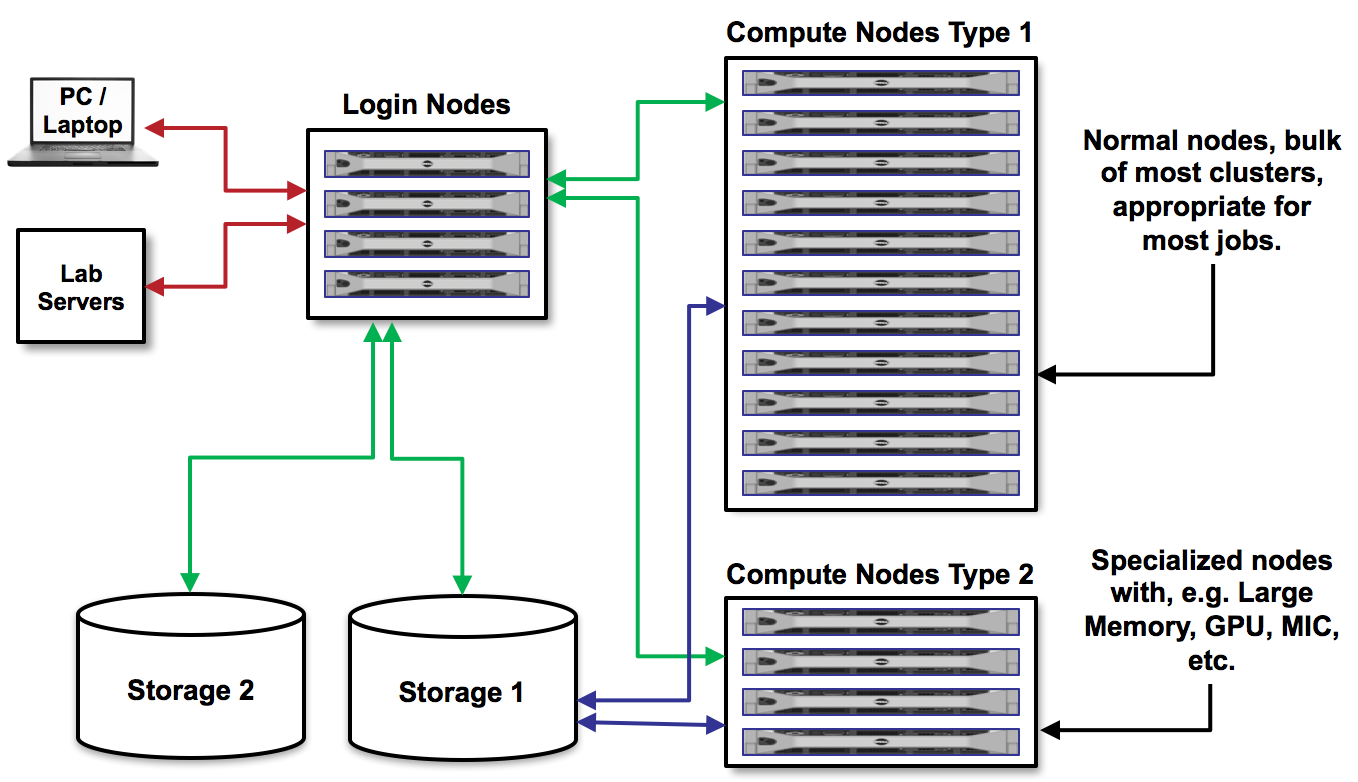
In this tutorial, we will use frontend nympha with an address nympha.metacentrum.cz for logging in. Nympha frontend runs on Debian 11 and has a home directory mounted on the storage plzen1 (accessible as /storage/plzen1/home/$USER/).
Note
Variable USER ($USER) is automatically set after log in and contains the real username.
Important
MetaCentrum for log in does not fully support traditional authentication with SSH keys. MetaCentum uses the Kerberos system for authentication, which requires a username and password.
Tip
You can install and configure Kerberos on your personal computer (available for all operating systems). It allows you to generate a local Kerberos ticket with a lifetime of up to 24 hours and log in to Metacentrum nodes without typing a password for this period.
Windows users can use (for example) an SSH client PuTTY (as described here). CLI users can open their terminals and type the following command (replace a string user_name with your actual MetaCentrum username) and then your password.
ssh user_name@nympha.metacentrum.czNote
No characters appear during the password typing in the terminal. This is a standard security behaviour.
Important
Do not share your MetaCentrum password with someone else. It could be interpreted as violating the terms of use, and your account could be banned.
When you see the overview of your quotas on individual storages, you have been successfully logged in. Expand the following section for comparison. The colour scheme will vary and is a matter of personal terminal settings.
Expand the following section to see what a successful login looks like.

In Metacentrum, we lack the visual interface typical for desktop computers. Instead, we use text commands. The following table shows a few commands for fundamental work with the Linux command line. Most of them we will use further during the course.
Tip
There are hundreds of other commands. You can find more comprehensive tutorials related to this topic online — for example, here.
| Command | Action |
|---|---|
pwd |
Shows the current working directory’s path. |
ls |
Lists a directory’s content. |
ls -lh |
Lists a directory’s content in column and human-readable format. |
mkdir |
Creates a new directory. |
cat |
Displays the content of a file. |
cd |
Changes the working directory. |
cp |
Copies files. |
cp -r |
Copies files and directories with their content. |
head |
Prints first 10 lines. |
mv |
Moves or renames files and directories. |
rm |
Removes a file. |
rmdir |
Removes an empty directory. |
rm -r |
Removes directory with its content. |
touch |
Creates a file without content. |
scp |
Transfers data. |
. or ./ |
Refers to the present location. |
.. or ../ |
Refers to the parent directory. |
After successful log in, you will be (by default) located in your home directory. You can execute the command pwd to print an absolute path of your position in the system.
pwdYour current location determines which files and directories you can see/access/use - which are located at the same position in the system. Execute the command ls to see what files and directories are available at this location, i.e., in your home directory.
lsWe can create a new directory named test_directory. If you do not like this name, you can choose a different one 😉
mkdir test_directoryWhen the folder test_directory was created, execute these two commands and compare what was printed in the terminal.
lsls -lNote
The terms folder and directory are interchangeable.
We will also need to edit text files (specifically the shell scripts for the batch jobs). We can use the touch command to create an empty file. Let's name this file as test_file.txt.
touch test_file.txtYou can execute the ls or ls -lh command again (depending on your preference) to check whether file test_file.txt was created successfully.
Expand this section and compare results with me.
(BULLSEYE)vorel@nympha:~$ ls
test_directory test_file.txt
(BULLSEYE)vorel@nympha:~$ ls -lh
total 512
drwxr-xr-x 2 vorel meta 4.0K Nov 28 15:21 test_directory
-rw-r--r-- 1 vorel meta 0 Nov 28 15:25 test_file.txtLet's use the text editor nano to open and further edit the text file test_file.txt.
nano test_file.txtTip
Use the tab key to autocomplete file or folder names.
Note
Metacentrum also offers other editors, such as vi, vim or mcedit.
You can start typing any text when the nano editor is open. When you are done, press the keys ctrl and x simultaneously. You will be asked if you want to save changes (Save modified buffer?). Press y, and as we want to overwrite the original file test_file.txt, press the enter key.
When you want to print the content of the text file in the terminal, try to use the command cat.
cat test_file.txtWarning
The cat command prints the entire content of a file in the terminal at once. Therefore, it is not a suitable way to view very long text files. More friendly is the less command, which shows the content of a text file by one page (one screen) at a time.
Finally, we can try to rename the file test_file.txt to test_file_renamed.txt and move it into the directory test_directory.
Note
Remember, the mv command can rename and move files and folders.
mv test_file.txt test_file_renamed.txtmv test_file_renamed.txt test_directoryIn the last step of this chapter, we can check whether the renamed file test_file_renamed.txt was moved to the correct position, and if yes, we can delete folder test_directory with its content because we will have no further use for it.
ls test_directoryrm -r test_directorySoftware tools available in MetaCentrum are accessible as environment modules. The system of modules is a concept simplifying the use of different software in a precise and controlled manner. Each program (in each version, eventually with additional modifications) is prepared as an individual module that must be activated (loaded) before use. The activation modifies the user's environment and sets everything necessary (especially variables $PATH and $LD_LIBRARY_PATH and loads dependencies) for the program's run.
Tip
How to work with MetaCentrum modules is described in detail here.
Note
MetaCentrum provides a few thousand modules. With this number of modules, manual control is no longer possible, and sometimes, not everything works without problems. If you encounter any malfunction, please let us know at meta@cesnet.cz.
We can execute a few commands and discuss how the module system works. Let's try to load a module for BLAST+.
module ava blastmodule ava *blast*module ava blast-plus/module add blast-plus/2.12.0-gccmodule listmodule rm blast-plusmodule listWarning
Remember that each activated module somehow modifies your environment. Loading many modules in one session can lead to conflicts and, as a result, to the non-functionality of some applications. The module that can be in conflict with others can be reliably limited only to defined parts of the job.
( module add module_name_1
programme ...
)
# After the right-round bracket, the used module is inactive, and all environmental modifications are suppressed.We will start this hands-on course by downloading the raw sequencing data from the NCBI Sequence Read Archive, followed by a quality check.
The methodology will include:
- starting the interactive job and navigating to the scratch directory.
- downloading the raw reads.
- quality control of Illumina and Oxford Nanopore raw reads.
- a visual assessment of produced graphs
Important
Scratch storage is a storage for temporary files and processed data of running jobs. This storage should be used only during computations and freed immediately after your job ends. The location of the scratch directory is defined by a system variable SCRATCHDIR.
First of all, we will submit an interactive job. The meaning of individual parts of the command is explained below.
qsub -I -l select=1:ncpus=2:mem=10gb:scratch_local=20gb -l walltime=2:00:00 -q MetaSeminar| Parameter | Action |
|---|---|
qsub |
Command that submits jobs. |
-I |
Declares that the job is to be run interactively. |
select=1 |
Reserves resources on one physical node. |
ncpus=2 |
Reserves two processors. |
mem=10gb |
Reserves 10 GB of RAM. |
scratch_local=20gb |
Reserves 20 GB of disk space on scratch. |
walltime=2:00:00 |
Reserves two hours for the job. |
-q MetaSeminar |
Submits job into queue MetaSeminar. |
After starting the job, go to the scratch directory, defined as the variable SCRATCHDIR.
cd $SCRATCHDIRImportant
Variable SCRATCHDIR ($SCRATCHDIR) is automatically set for each job. Always use the $SCRATCHDIR. The real path to the scratch directory is unknown before the start of the job because it contains an assigned job number. For example, /scratch/user_name/job_123456789.meta-pbs.metacentrum.cz.
For data download, we will use the SRA Toolkit (version 3.0.3), which is a tool developed by NCBI for SRA data manipulation. In MetaCentrum, this tool is available as a Conda environment.
module add conda-modules
conda activate sra-tools-3.0.3
fasterq-dump -e 2 -p -x SRR24321377 SRR24321378| Flag | Meaning |
|---|---|
-e 2 |
Uses two threads. |
-p |
Shows progress. |
-x |
Prints more details. |
SRR24321377 |
Oxford Nanopore reads. |
SRR24321378 |
Illumina paired-end reads. |
Important
Oxford Nanopore sequencers produce data in fast5 format that contains additional information besides sequence data. In this tutorial, we download data already converted into fastq format.
We can check the content of the scratch directory via the ls -lh command. Do not use the cat command to explore the content of individual fastq files!
We can also print out the first ten lines from each file, check the data visually and count the number of sequences in each file.
head SRR24321377.fastq
head SRR24321378_*.fastq
for NAME in SRR24321377.fastq SRR24321378_1.fastq SRR24321378_2.fastq; do wc -l $NAME | awk '{print $1/4}'; doneAnd we can rename the downloaded files for better clarity.
mv SRR24321377.fastq ONT_raw_SRR24321377.fastq
mv SRR24321378_1.fastq Illumina_raw_SRR24321378_1.fastq
mv SRR24321378_2.fastq Illumina_raw_SRR24321378_2.fastqWe no longer need activated environment sra-tools-3.0.3 or module conda-modules, so we can deactivate them.
conda deactivate
module rm conda-modulesLet's continue with the FastQC utility to check the quality of Illumina raw reads. Try to execute the command module ava fastqc/. What is the latest FastQC version installed in MetaCentrum?
Important
MetaCentrum users are allowed to install software tools on their own, preferably in their home directories. We fully support these activities. Our only condition is that there is no violation of the license rights of the given tool or the MetaCentrum terms and conditions.
A newer version of FastQC is available. FastQC is distributed as a set of scripts and Java jar files. No compilation is needed. Thus, we will download, extract and use the latest version of FastQC. It is pretty simple, and everyone can make it 😊
wget https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.12.1.zip
unzip fastqc_v0.12.1.zip
ls
ls FastQC/
ls -lh FastQC | grep fastqc
chmod u+x FastQC/fastqc
module add openjdk
FastQC/fastqc -h
FastQC/fastqc -t 2 Illumina_raw_SRR24321378_*
ls -lh
module rm openjdk| Command/flag | Meaning |
|---|---|
wget |
Downloads files from the web. |
unzip |
Extract files from a ZIP archive. |
| |
Combines two commands. |
grep |
Searches matching text. |
chmod u+x |
Permits the user to execute a file. |
openjdk |
Module for open source java. |
-t 2 |
Uses two threads. |
Statistic graphs produced by FastQC are saved as html files, which can be downloaded and opened in a web browser on your local computer. Before proceeding, we will also perform quality control for Oxford Nanopore reads. We will use the NanoPlot tool, which can be installed through a Mamba package manager. In MetaCentrum, it is available as a module called mambaforge.
Note
Conda and Mamba package managers are very popular tools which allow fast and fully automated installations of various software. Each software is installed in a separate environment to avoid conflicts with other tools. In MetaCentrum, Mamba is preferred over Conda because it is designed to be faster and more efficient. Installations through Mamba are mostly straightforward and non-problematic.
module add mambaforge
mamba create -p /storage/plzen1/home/$USER/nanoplot-env -c bioconda nanoplot -y
mamba activate /storage/plzen1/home/$USER/nanoplot-env
NanoPlot --help
NanoPlot -t 2 -o ont_outdir -c red --plots dot --N50 --fastq ONT_raw_SRR24321377.fastq
mamba deactivate && mamba deactivate| Command/flag | Meaning |
|---|---|
mamba create -p... |
Crates a new environment in a specified location and installs a NanoPlot package from the Bioconda channel. |
-y |
Automatically approves every action. |
mamba activate |
Activates existing environment. |
-t 2 |
Uses two threads. |
-o |
Sets the name of the directory with results. |
-c |
Sets the colour of produced graphs. |
--plots |
Specifies the plot style. |
--50 |
Shows N50 mark. |
--fastq |
Input file in a fastq format. |
&& |
Executes the second command when the first one ends successfully. |
Finally, we can move results from the scratch directory to the home directory (/storage/plzen1/home/$USER) for further examination.
cp Illumina_raw_SRR24321378_*.fastq /storage/plzen1/home/$USER
cp ONT_raw_SRR24321377.fastq /storage/plzen1/home/$USER
cp Illumina_raw_SRR24321378_*_fastqc.html /storage/plzen1/home/$USER
cp -r ont_outdir /storage/plzen1/home/$USERWe no longer need the remaining content of the scratch directory. So, we call the clean_scratch utility, which will remove all remaining data. Then, we execute the exit command to quit the running interactive job.
clean_scratch
exitImportant
Scratch directories are not backed up! Once data is deleted, it is not possible to restore it.
From the previous chapter, we have two types of data (reads and files with quality information), and we will use them as examples for:
- download the data from MetaCentrum to the local computer.
- upload the data to MetaCentrum.
- transfer the data between storages.
- archive and backup the data.
Important
How to effectively manipulate the data is comprehensively described here.
Tip
It is a good practice to compress larger data volumes (into .zip, .gz, .tar.gz, etc.) before manipulation.
Firstly, we will download the results from the quality check step. This means we will download the data from the MetaCentrum storage plzen1 to the local computer.
In general, small files and folders can be downloaded/uploaded through the frontend servers. For bigger volumes of data, it is recommended to access storage servers directly. A list of all MetaCentrum storage servers is deposited here.
The easiest way to download data from the remote server is via a terminal. Let's execute a few commands:
scp user_name@storage-plzen1.metacentrum.cz:Illumina_raw_SRR24321378_\*_fastqc.html .
# alternatively:
scp user_name@nympha.metacentrum.cz:Illumina_raw_SRR24321378_\*_fastqc.html .scp -r user_name@storage-plzen1.metacentrum.cz:ont_outdir .
# alternatively:
scp -r user_name@nympha.metacentrum.cz:ont_outdir .scp user_name@storage-plzen1.metacentrum.cz:ONT_raw_SRR24321377.fastq .
# alternatively:
scp user_name@nympha.metacentrum.cz:ONT_raw_SRR24321377.fastq .General syntax with path is scp user_name@server_name:/path/to/any/file/ /path/where/to/save/it/on/my/computer. scp is a traditional Linux command with many tutorials on how to use it.
Important
The directory structure on NFS4 storages and frontend servers is not identical!
$ ssh vorel@nympha.metacentrum.cz
$ pwd
/storage/plzen1/home/vorel
$ ls
Illumina_raw_SRR24321378_1.fastq Illumina_raw_SRR24321378_2.fastq
$ sftp vorel@storage-plzen1.metacentrum.cz
$ pwd
Remote working directory: /home/vorel
Illumina_raw_SRR24321378_1.fastq Illumina_raw_SRR24321378_1_fastqc.html
Some users prefer (or need) graphical FTP/SFTP clients for interactive access. Such clients are, for example, WinSCP, FileZilla or CyberDuck. All these clients need to be correctly configured before use. To access the MetaCentrum, do the following:
- select FTP or SFTP protocol
- set port
22 - insert your username and password
- server address (use
nympha.metacentrum.czin this tutorial)
An example below shows a configuration of CyberDuck to access the nympha frontend. The access point is, by default, set as a home directory (/storage/plzen1/home/$USER/).

After the configuration, you can manually download the same files and folder as we did before through the terminal.
Important
Finally, we can explore the downloaded files and evaluate the quality of raw reads...
Data upload for users of FTP/SFTP clients works as easily as download. They mainly work with the Drag-and-drop technique or the Upload option. The scp command also works similarly, only destinations are switched. In general: scp /path/to/any/folder/or/file/on/my/computer user_name@server_name:/path/where/to/save/it.
mv ONT_raw_SRR24321377.fastq ONT_raw_SRR24321377.fastq_renamed
scp /local/path/ONT_raw_SRR24321377.fastq_renamed user_name@storage-plzen1.metacentrum.cz:
# alternatively:
scp /local/path/ONT_raw_SRR24321377.fastq_renamed user_name@nympha.metacentrum.cz:Tip
For a copy of a folder through scp, it is necessary to use the -r flag. When the folder is compressed (for example, folder.gz), the -r flag is not required because the compressed file is handled as a single file.
Warning
Although you do not need to specify the path where to save something on the remote server (in the scp command), the colon character : after the server name is still required.
scp -r /local/path/folder user_name@storage-plzen1.metacentrum.cz:
Tip
Before transferring big data (a few TBs), you can email MetaCentrum user support (meta@cesnet.cz) and discuss the optimal strategy. Alternatively, we can transfer data for you.
Small data volumes can be transferred between storages directly from frontends by traditional commands cp and mv.
cp /storage/plzen1/home/$USER/ONT_raw_SRR24321377.fastq /storage/brno12-cerit/home/$USER
ls /storage/brno12-cerit/home/$USER
rm /storage/brno12-cerit/home/$USERThe same strategy can be applied for data transfer to and from the scratch directory within interactive and batch jobs.
cp /storage/plzen1/home/$USER/ONT_raw_SRR24321377.fastq $SCRATCHDIR
mv $SCRATCHDIR/output.txt /storage/plzen1/home/$USERIs it more suitable to transfer large volumes of data by the scp command from/to/between individual NFS4 storages. Let's try to solve a few examples...
scp /storage/plzen1/home/$USER/ONT_raw_SRR24321377.fastq storage-brno12-cerit.metacentrum.cz:~
ls /storage/brno12-cerit/home/$USERscp -r /storage/plzen1/home/$USER/ont_outdir storage-brno12-cerit.metacentrum.cz:~
ls /storage/brno12-cerit/home/$USERcd /storage/plzen1/home/$USER/
scp ONT_raw_SRR24321377.fastq storage-brno12-cerit.metacentrum.cz:~scp storage-brno12-cerit.metacentrum.cz:~/ONT_raw_SRR24321377.fastq /storage/plzen1/home/$USER/scp storage-plzen1.metacentrum.cz:~/ONT_raw_SRR24321377.fastq storage-brno12-cerit.metacentrum.cz:~Note
MetaCentrum storages are dedicated to data in active use. Unnecessary data of permanent value should be archived in a suitable place.
Important
MetaCentrum storages are not backed up on other servers. Data on storage is backed up on the same server in the form of snapshots. The snapshots are kept at least 14 days backwards. In the event of a catastrophic storage failure, data may be irretrievably lost.
MetaCentrum users can use additional storage capacity provided by the CESNET Storage Department. This storage capacity represents a simple way to back up and archive valuable data. The data is kept in several physical copies, so there is no risk of losing it due to a hardware failure.
Two special storages are mounted as:
/storage/du-cesnet/home/$USER/VO_metacentrum-tape_tape-archive- this archive storage keeps data "forever"./storage/du-cesnet/home/$USER/VO_metacentrum-tape_tape- backup storage with a life cycle of 12 months.
Important
Do not leave data in the home directory of this storage, i.e. in /storage/du-cesnet/home/$USER/. The data quota here is set very low (~50 MB).
Important
As on most other storages, there are also applied quotas for the volume of data and the total number of files. Quotas can be reviewed in the PBSmon.
Storages for archiving and backuping are accessible as other MetaCentrum storage. Try to execute the following examples:
cp ONT_raw_SRR24321377.fastq /storage/du-cesnet/home/$USER/VO_metacentrum-tape_tape-archive/scp ONT_raw_SRR24321377.fastq storage-du-cesnet.metacentrum.cz:~/VO_metacentrum-tape_tape-archiveWarning
The default home directory on the server storage-du-cesnet.metacentrum.cz is set as /storage/du-cesnet/home/$USER/. And it is not allowed to write to this home directory. Always specify a path ~/VO_metacentrum-tape_tape-archive or ~/VO_metacentrum-tape_tape when you use the scp command.
scp storage-plzen1.metacentrum.cz:~/ONT_raw_SRR24321377.fastq storage-du-cesnet.metacentrum.cz:~/VO_metacentrum-tape_tape-archivescp storage-du-cesnet.metacentrum.cz:~/VO_metacentrum-tape_tape-archive/ONT_raw_SRR24321377.fastq storage-plzen1.metacentrum.czNote
Storage du-cesnet is based on different technology and is significantly slower than other MetaCentrum storages. For that reason, it is recommended:
- archive/backup compressed files.
- use faster approaches than
cpormvcommands for data upload. - do not use this storage for calculations.
- if you need to search or decompress a large archive, create a copy on another (faster) MetaCentra storage.
Tip
Access with WinSCP or FileZilla can be configured based on this tutorial, respective this one. Server address is ftp.du4.cesnet.cz.
In the final chapter of this course, we will do a genome assembly from previously downloaded data. As we decided, we will use unprocessed raw reads for our humble testing purposes.
Create a new and empty file in your home directory (preferably /storage/plzen1/home/$USER).
touch run_batch_assembly.shOpen this file in the nano text editor as nano run_batch_assembly.sh and paste the entire content of the following script into the text editor.
#!/bin/bash
#PBS -q MetaSeminar
#PBS -l walltime=01:00:00
#PBS -l select=1:ncpus=2:mem=15gb:scratch_local=20gb
#PBS -N Batch_test_assembly
# test if a scratch directory exists
test -n "$SCRATCHDIR" || { echo >&2 "Variable SCRATCHDIR is not set!"; exit 1; }
# set a DATADIR variable
DATADIR=/storage/plzen1/home/$USER
# copy input files to the scratch directory
cp $DATADIR/Illumina_raw_SRR24321378_*.fastq $SCRATCHDIR || { echo >&2 "Error while copying Illumina input files!"; exit 2; }
cp $DATADIR/ONT_raw_SRR24321377.fastq $SCRATCHDIR || { echo >&2 "Error while copying ONT input file!"; exit 2; }
# go to the scratch directory
cd $SCRATCHDIR
head -n 4000000 Illumina_raw_SRR24321378_1.fastq > Illumina_raw_SRR24321378_1_partial.fastq
head -n 4000000 Illumina_raw_SRR24321378_2.fastq > Illumina_raw_SRR24321378_2_partial.fastq
head -n 10000 ONT_raw_SRR24321377.fastq > ONT_raw_SRR24321377_partial.fastq
# load a module for your application
module add unicycler/0.5.0
# run the calculation
unicycler-runner.py -1 Illumina_raw_SRR24321378_1_partial.fastq -2 Illumina_raw_SRR24321378_2_partial.fastq \
-l ONT_raw_SRR24321377_partial.fastq -o out_directory -t 2 --kmers 21 --kmer_count 12
# move results
mv out_directory/assembly.fasta $DATADIR || { echo >&2 "Result file(s) copying failed (with a code $?) !!"; exit 4; }
# clean the scratch directory
clean_scratchAs we are limited by time and hardware resources dedicated to this course, we will subsample Illumina reads on 1,000,000 pairs and Oxford Nanopore reads on 2,500 reads to reduce the run time. The only desired output is the fasta file assembly.fasta with the assembled genome.
Caution
A subsampling technique used in the batch script (head -n 4000000...) is wrong and should never be used for serious processing of scientific data.
Note
With the current setting, the expected runtime is ~30 minutes.
The prepared script can be submitted to the scheduling system.
qsub run_batch_assembly.shIn the final step, we will compare the basic characteristics of our assembly with the published one. In the interactive job, we will use the QUAST tool; the procedure is following.
qsub -I -l select=1:ncpus=2:mem=10gb:scratch_local=20gb -l walltime=1:00:00 -q MetaSeminar
cd $SCRATCHDIR
cp /storage/plzen1/home/$USER/assembly.fasta .
module add conda-modules
conda activate quast_v5.2.0_py3.9
quast -o out_dir -t 2 --fast assembly.fasta
cat out_dir/report.txt
clean_scratch
exit