
- Graph Embedding:深度学习推荐系统的"基本操作"
- The Power of Graphs in Machine Learning and Sequential Decision-Making
- http://www.ai3sd.org/
- https://heidelberg.ai/2019/07/09/graph-neural-networks.html
- https://sites.google.com/site/rdftestxyz/home
- Lecture 11: Learning on Non-Euclidean Domains
- Network Embedding
- 如何评价百度新发布的NLP预训练模型ERNIE? - 知乎
- What Can Neural Networks Reason About?
- Deep Geometric Matrix Completion by Federico Monti
- Jure Leskovec.
- http://www-connex.lip6.fr/~denoyer/wordpress/
- https://blog.feedly.com/learning-context-with-item2vec/
- Computational Learning and Memory Group
- Beyond deep learning
- Cognitive Computation Group @ U. Penn.
- Computational cognitive modeling
- Mechanisms of geometric cognition
- Computational Cognitive Science Lab
- https://jian-tang.com/teaching/graph2019
- Introducing Grakn & Knowledge Graph Convolutional Networks: Dec 4, 2018 · Paris, France
- International Workshop on Deep Learning for Graphs and Structured Data Embedding
- HYPERBOLIC DEEP LEARNING: A nascent and promising field
Images are stored in computer as matrix roughly. The spatial distribution of pixel on the screen project to us a colorful digitalized world.
Convolutional neural network(ConvNet or CNN) has been proved to be efficient to process and analyses the images for visual cognitive tasks.
What if we generalize these methods to graph structure which can be represented as adjacent matrix?
| Image | Graph |
|---|---|
| Convolutional Neural Network | Graph Convolution Network |
| Attention | Graph Attention |
| Gated | Gated Graph Network |
| Generative | Generative Models for Graphs |
Advanced proceedings of natural language processing(NLP) shone a light into semantic embedding as one potential approach to knowledge representation.
The text or symbol, strings in computer, is designed for natural people to communicate and understand based on the context or situation, i.e., the connections of entities and concepts are essential.
What if we generalize these methods to connected data?
Graph embedding, preprocessing of graph data processing, is an example of representation learning to find proper numerical representation form of graph data structure.
It maps the graph structure to numerical domain:
- https://github.com/thunlp/NRLpapers
- https://github.com/thunlp/GNNPapers
- http://snap.stanford.edu/proj/embeddings-www/
- https://arxiv.org/abs/1709.05584
- http://cazabetremy.fr/Teaching/EmbeddingClass.html
- Awesome Graph Embedding
- A Beginner's Guide to Graph Analytics and Deep Learning
- Representation Learning on Graphs: Methods and Applications
- DOOCN-XII: Network Representation Learning Dynamics On and Of Complex Networks 2019
- 15TH INTERNATIONAL WORKSHOP ON MINING AND LEARNING WITH GRAPHS
- Hyperbolic geometry and real life networks
- Network Embedding as Matrix Factorization: Unifying DeepWalk, LINE, PTE, and node2vec
- Representation Learning on Networks
DeepWalk is an approach for learning latent representations of vertices in a network, which maps the nodes in the graph into real vectors:
$$
f: \mathbb{V}\to\mathbb{R}^{d}.
$$
DeepWalk generalizes recent advancements in language modeling and unsupervised feature learning (or deep learning) from sequences of words to graphs. DeepWalk uses local information obtained from truncated random walks to learn latent representations by treating walks as the equivalent of sentences.
And if we consider the text as digraph, word2vec is an specific example of DeepWalk.
Given the word sequence
- DeepWalk
$G, w, d, \gamma, t$
- Input: graph
$G(V, E)$ ; window size$w$ ; embedding size$d$ ; walks per vertex$\gamma$ ; walk length$t$ .- Output: matrix of vertex representations
$\Phi\in\mathbb{R}^{|V|\times d}$
- Initialization: Sample
$\Phi$ from$\mathbb{U}^{|V|\times d}$ ;
- Build a binary Tree T from V;
- for
$i = 0$ to$\gamma$ do
$O = Shuffle(V )$ - for each
$v_i \in O$ do$W_{v_i}== RandomWalk(G, v_i, t)$ $SkipGram(Φ, W_{v_i}, w)$ - end for
- end for
$SkipGram(Φ, W_{v_i}, w)$
-
- for each
$v_j \in W_{v_i}$ do
-
- for each
$u_k \in W_{v_i}[j - w : j + w]$ do
- for each
-
$J(\Phi)=-\log Pr(u_k\mid \Phi(v_j))$
-
$\Phi =\Phi -\alpha\frac{\partial J}{\partial \Phi}$
-
- end for
- for each
-
- end for
Computing the partition function (normalization factor) is expensive, so instead we will factorize the
conditional probability using Hierarchical softmax.
If the path to vertex
Now,
- DeepWalk: Online Learning of Social Representations
- DeepWalk at github
- Deep Walk Project @perozzi.net
- http://www.cnblogs.com/lavi/p/4323691.html
- https://www.ijcai.org/Proceedings/16/Papers/547.pdf
node2vec is an algorithmic framework for representational learning on graphs. Given any graph, it can learn continuous feature representations for the nodes, which can then be used for various downstream machine learning tasks.
By extending the Skip-gram architecture to networks, it seeks to optimize the following objective function,
which maximizes the log-probability of observing a network neighborhood
Conditional independence and Symmetry in feature space are expected to make the optimization problem tractable.
We model the conditional likelihood of every source-neighborhood node pair as a softmax
unit parametrized by a dot product of their features:
$$
Pr(n_i\mid f(u))=\frac{\exp(f(n_i)\cdot f(u))}{\sum_{v\in V} \exp(f(v)\cdot f(u))}.
$$
The objective function simplifies to
- https://zhuanlan.zhihu.com/p/30599602
- http://snap.stanford.edu/node2vec/
- https://cs.stanford.edu/~jure/pubs/node2vec-kdd16.pdf
- https://www.kdd.org/kdd2016/papers/files/rfp0218-groverA.pdf
This is a paper about identifying nodes in graphs that play a similar role based solely on the structure of the graph, for example computing the structural identity of individuals in social networks. That’s nice and all that, but what I personally find most interesting about the paper is the meta-process by which the authors go about learning the latent distributed vectors that capture the thing they’re interested in (structural similarity in this case). Once you’ve got those vectors, you can do vector arithmetic, distance calculations, use them in classifiers and other higher-level learning tasks and so on. As word2vec places semantically similar words close together in space, so we want structurally similar nodes to be close together in space.
Struc2vec has four main steps:
- Determine the structural similarity between each vertex pair in the graph, for different neighborhood sizes.
- Construct a weighted multi-layer graph, in which each layer corresponds to a level in a hierarchy measuring structural similarity (think: ‘at this level of zoom, these things look kind of similar�?).
- Use the multi-layer graph to generate context for each node based on biased random walking.
- Apply standard techniques to learn a latent representation from the context given by the sequence of nodes in the random walks.

- STRUC2VEC(图结构→向量)论文方法解读
- struc2vec: Learning Node Representations from Structural Identity
- https://arxiv.org/abs/1704.03165
- Struc2vec: learning node representations from structural identity
word2vec
In natural language processing, the word can be regarded as the node in a graph, which only takes the relation of locality or context.
It is difficult to learn the concepts or the meaning of words. The word embedding technique word2vec maps the words to fixed length real vectors:
$$
f: \mathbb{W}\to\mathbb{V}^d\subset \mathbb{R}^d.
$$
The skip-gram model assumes that a word can be used to generate the words that surround it in a text sequence.
We assume that, given the central target word, the context words are generated independently of each other.
The conditional probability of generating the context word for the given central target word can be obtained by performing a softmax operation on the vector inner product: $$ P(w_o|w_c) = \frac{\exp(u_o^T u_c)}{\sum_{i\in\mathbb{V}} \exp(u_i^T u_c)}, $$
where vocabulary index set
Here, any time step that is less than 1 or greater than
The skip-gram model parameters are the central target word vector and context word vector for each individual word. In the training process, we are going to learn the model parameters by maximizing the likelihood function, which is also known as maximum likelihood estimation. his is equivalent to minimizing the following loss function: $$ -\log(\prod_{t=1}^{T}\prod_{-m\leq j \leq m, j\not = i}{P}(w^{(t+j)}\mid w^{(j)})) = \ -\sum_{t=1}^{T}\sum_{-m\leq j \leq m, j \not= i} \log({P}(w^{(t+j)}|w^{(j)}))). $$
And we could compute the negative logarithm of the conditional probability $$ -\log(P(w_o|w_c)) = -\log(\frac{\exp(u_o^T u_c)}{\sum_{i\in\mathbb{V}} \exp(u_i^T u_c)}) \= -u_o^T u_c + \log(\sum_{i\in\mathbb{V}} \exp(u_i^T u_c)). $$
Then we could compute the gradient or Hessian matrix of the loss functions to update the parameters such as:
The continuous bag of words (CBOW) model is similar to the skip-gram model. The biggest difference is that the CBOW model assumes that the central target word is generated based on the context words before and after it in the text sequence. Let central target word
- https://code.google.com/archive/p/word2vec/
- https://skymind.ai/wiki/word2vec
- https://arxiv.org/abs/1402.3722v1
- https://zhuanlan.zhihu.com/p/35500923
- https://zhuanlan.zhihu.com/p/26306795
- https://zhuanlan.zhihu.com/p/56382372
- http://anotherdatum.com/vae-moe.html
- https://d2l.ai/chapter_natural-language-processing/word2vec.html
- https://www.gavagai.io/text-analytics/word-embeddings/
Doc2Vec
- https://blog.csdn.net/Walker_Hao/article/details/78995591
- Distributed Representations of Sentences and Documents
- Sentiment Analysis using Doc2Vec
- Learning and Reasoning with Graph-Structured Representations, ICML 2019 Workshop
- Transformer结构及其应用--GPT、BERT、MT-DNN、GPT-2 - Ph0en1x的文�? - 知乎
- 放弃幻想,全面拥抱Transformer:自然语言处理三大特征抽取器(CNN/RNN/TF)比较
- Statistical Models of Language
- Semantic Word Embeddings
- Word Embeddings
- GloVe: Global Vectors for Word Representation Jeffrey Pennington, Richard Socher, Christopher D. Manning
- BERT-is-All-You-Need
- Word embedding
- Open Sourcing BERT: State-of-the-Art Pre-training for Natural Language Processing? Friday, November 2, 2018
- The Illustrated BERT, ELMo, and co. (How NLP Cracked Transfer Learning)
- Deep Semantic Embedding
- 无监督词向量/句向量?:W2v/Glove/Swivel/ELMo/BERT
- The Expressive Power of Word Embeddings
Gradient Boosted Categorical Embedding and Numerical Trees (GB-CSENT) is to combine Tree-based Models and Matrix-based Embedding Models in order to handle numerical features and large-cardinality categorical features.
A prediction is based on:
- Bias terms from each categorical feature.
- Dot-product of embedding features of two categorical features,e.g., user-side v.s. item-side.
- Per-categorical decision trees based on numerical features ensemble of numerical decision trees where each tree is based on one categorical feature.
In details, it is as following: $$ \hat{y}(x) = \underbrace{\underbrace{\sum_{i=0}^{k} w_{a_i}}{bias} + \underbrace{(\sum{a_i\in U(a)} Q_{a_i})^{T}(\sum_{a_i\in I(a)} Q_{a_i}) }{factors}}{CAT-E} + \underbrace{\sum_{i=0}^{k} T_{a_i}(b)}_{CAT-NT}. $$ And it is decomposed as the following table.
| Ingredients | Formulae | Features |
|---|---|---|
| Factorization Machines | $\underbrace{\underbrace{\sum_{i=0}^{k} w_{a_i}}{bias} + \underbrace{(\sum{a_i\in U(a)} Q_{a_i})^{T}(\sum_{a_i\in I(a)} Q_{a_i}) }{factors}}{CAT-E}$ | Categorical Features |
| GBDT | Numerical Features |
- http://www.hongliangjie.com/talks/GB-CENT_SD_2017-02-22.pdf
- http://www.hongliangjie.com/talks/GB-CENT_SantaClara_2017-03-28.pdf
- http://www.hongliangjie.com/talks/GB-CENT_Lehigh_2017-04-12.pdf
- http://www.hongliangjie.com/talks/GB-CENT_PopUp_2017-06-14.pdf
- http://www.hongliangjie.com/talks/GB-CENT_CAS_2017-06-23.pdf
- http://www.hongliangjie.com/talks/GB-CENT_Boston_2017-09-07.pdf
- Talk: Gradient Boosted Categorical Embedding and Numerical Trees
- Paper: Gradient Boosted Categorical Embedding and Numerical Trees
- https://qzhao2018.github.io/zhao/
Gaussian Auto Embeddings
http://koaning.io/gaussian-auto-embeddings.html
Atom2Vec
M. V. Diudea, I. Gutman and L. Jantschi wrote in the preface of the book Molecular Topology: One of the principal goals of chemistry is to establish (causal) relations between the chemical and physical (experimentally observable and measurable) properties of substance and the structure of the corresponding molecules. Countless results along these lines have been obtained, and their presentation comprise significant parts of textbooks of organic, inorganic and physical chemistry, not to mention treatises on theoretical chemistry
tile2Vec
- RDF2Vec: RDF Graph Embeddings and Their Applications
- EmbedS: Scalable and Semantic-Aware Knowledge Graph Embeddings
graph2vec
graph2vec is to learn data-driven distributed representations of arbitrary sized graphs in an unsupervised manner and are task agnostic.
- graph2vec: Learning Distributed Representations of Graphs
- https://allentran.github.io/graph2vec
- http://humanativaspa.it/tag/graph2vec/
- https://zhuanlan.zhihu.com/p/33732033
- Awesome graph embedding
- Graph Embedding Methods
- Graph Embedding @ deep learning pattern
- Representation learning in graph and manifold
- Learning and Reasoning with Graph-Structured Representations, ICML 2019 Workshop
- LINE: Large-scale Information Network Embedding
- Latent Network Summarization: Bridging Network Embedding and Summarization
- NodeSketch: Highly-Efficient Graph Embeddings via Recursive Sketching
- GEMS Lab
- Graph Embeddings — The Summary
- Graph Embeddings search result @ Arxiv-sanity
- http://smir2014.noahlab.com.hk/paper%204.pdf
- Deep Embedding Logistic Regression
- DeViSE: A Deep Visual-Semantic Embedding Model
- Deep Visual-Semantic Alignments for Generating Image Descriptions
- Semantic Embedding for Sketch-Based 3D Shape Retrieval
- Spherical and Hyperbolic Embeddings of Data
- Embedding Networks in Hyperbolic Spaces
- Characterizing the analogy between hyperbolic embedding and community structure of complex networks
- Poincaré Embeddings for Learning Hierarchical Representations
- Implementing Poincaré Embeddings
- Hyperbolic Embedding search result @Arxiv-sanity

- Hyperbolic Embeddings with a Hopefully Right Amount of Hyperbole
- HyperE: Hyperbolic Embeddings for Entities
- Efficient embedding of complex networks to hyperbolic space via their Laplacian
- Embedding Text in Hyperbolic Spaces
- Hyperbolic Function Embedding: Learning Hierarchical Representation for Functions of Source Code in Hyperbolic Spaces
- http://hyperbolicdeeplearning.com/papers/
| --- | --- |
|---|---|
 |
 |
- https://zhuanlan.zhihu.com/p/47489505
- http://blog.lcyown.cn/2018/04/30/graphencoding/
- https://blog.csdn.net/NockinOnHeavensDoor/article/details/80661180
- http://building-babylon.net/2018/04/10/graph-embeddings-in-hyperbolic-space/
- https://paperswithcode.com/task/graph-embedding
If images is regarded as a matrix in computer and text as a chain( or sequence), their representation contain all the spatial and semantic information of the entity.
Graph can be represented as adjacency matrix as shown in Graph Algorithm. However, the adjacency matrix only describe the connections between the nodes.
The feature of the nodes does not appear. The node itself really matters.
For example, the chemical bonds can be represented as adjacency matrix while the atoms in molecule really determine the properties of the molecule.
A simple and direct way is to concatenate the feature matrix adjacency matrix
And what is the output? How can deep learning apply to them? And how can we extend the tree-based algorithms such as decision tree into graph-based algorithms?
For these models, the goal is then to learn a function of signals/features on a graph
$G=(V,E)$ which takes as input:
- A feature description
$x_i$ for every node$i$ ; summarized in a$N\times D$ feature matrix${X}$ ($N$ : number of nodes,$D$ : number of input features);- A representative description of the graph structure in matrix form; typically in the form of an adjacency matrix
${A}$ (or some function thereof)
and produces a node-level output
$Z$ (an$N\times F$ feature matrix, where$F$ is the number of output features per node). Graph-level outputs can be modeled by introducing some form of pooling operation (see, e.g. Duvenaud et al., NIPS 2015).
Every neural network layer can then be written as a non-linear function
$$
{H}{i+1} = \sigma \circ ({H}{i}, A)
$$
with ${H}0 = {X}{in}$ and
For example, we can consider a simple form of a layer-wise propagation rule
$$
{H}{i+1} = \sigma \circ ({H}{i}, A)=\sigma \circ(A {H}{i} {W}{i})
$$
where
-
But first, let us address two limitations of this simple model: multiplication with
$A$ means that, for every node, we sum up all the feature vectors of all neighboring nodes but not the node itself (unless there are self-loops in the graph). We can "fix" this by enforcing self-loops in the graph: we simply add the identity matrix$I$ to$A$ . -
The second major limitation is that
$A$ is typically not normalized and therefore the multiplication with$A$ will completely change the scale of the feature vectors (we can understand that by looking at the eigenvalues of$A$ ).Normalizing${A}$ such that all rows sum to one, i.e.$D^{-1}A$ , where$D$ is the diagonal node degree matrix, gets rid of this problem.
In fact, the propagation rule introduced in Kipf & Welling (ICLR 2017) is given by:
$$
{H}{i+1} = \sigma \circ ({H}{i}, A)=\sigma \circ(\hat{D}^{-\frac{1}{2}} \hat{A} \hat{D}^{-\frac{1}{2}} {H}{i} {W}{i}),
$$
with
Like other neural network, GCN is also composite of linear and nonlinear mapping. In details,
-
$\hat{D}^{-\frac{1}{2}} \hat{A} \hat{D}^{-\frac{1}{2}}$ is to normalize the graph structure; - the next step is to multiply node properties and weights;
- Add nonlinearities by activation function
$\sigma$ .
See more at experoinc.com or https://tkipf.github.io/.
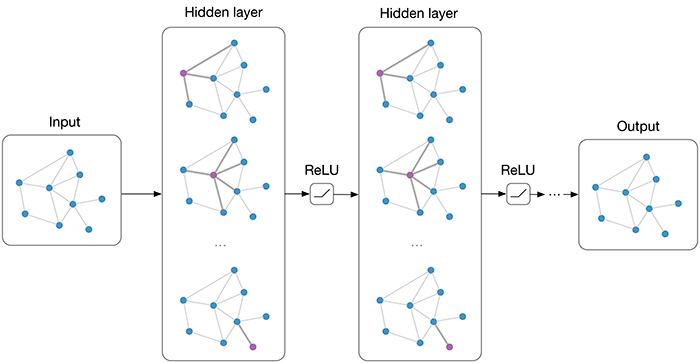

GCN can be regarded as the counterpart of CNN for graphs so that the optimization techniques such as normalization, attention mechanism and even the adversarial version can be extended to the graph structure.
- Node Classification by Graph Convolutional Network
- GRAPH CONVOLUTIONAL NETWORKS
- https://benevolent.ai/publications
Compositional layers of convolutional neural network can be expressed as
$$ \hat{H}{i} = P\oplus H{i-1} \ \tilde{H_i} = C_i\otimes(\hat{H}_{t}) \ Z_i = \mathrm{N}\cdot \tilde{H_i} \ H_i = Pooling\cdot (\sigma\circ Z_i) $$
where
Xavier Bresson gave a talk on New Deep Learning Techniques FEBRUARY 5-9, 2018. We would ideally like our graph convolutional layer to have:
- Computational and storage efficiency (requiring no more than
$O(E+V)$ time and memory); - Fixed number of parameters (independent of input graph size);
- Localisation (acting on a local neighbourhood of a node);
- Ability to specify arbitrary importances to different neighbours;
- Applicability to inductive problems (arbitrary, unseen graph structures).
| CNN | GCN | --- |
|---|---|---|
| padding | ? | ? |
| convolution | ? | Information of neighbors |
| pooling | ? | Invariance |
-
Spectral graph theoryallows to redefine convolution in the context of graphs with Fourier analysis. - Graph downsampling
$\iff$ graph coarsening$\iff$ graph partitioning: Decompose${G}$ into smaller meaningful clusters. - Structured pooling: Arrangement of the node indexing such that adjacent nodes are hierarchically merged at the next coarser level.
Laplacian operator is represented as a positive semi-definite
| Laplacian | Representation |
|---|---|
| Unnormalized Laplacian | |
| Normalized Laplacian | |
| Random walk Laplacian | |
|
|
|
Eigendecomposition of graph Laplacian:
where
Convolution of two vectors
where the last equation is because the matrix multiplication is associative and
Graph Convolution: Recursive Computation with Shared Parameters:
- Represent each node based on its neighbourhood
- Recursively compute the state of each node by propagating previous states using relation specific transformations
- Backpropagation through Structure
Vanilla spectral graph ConvNets
Every graph convolutional layer starts off with a shared node-wise feature transformation (in order to achieve a higher-level representation), specified by a weight matrix
In general, to satisfy the localization property, we will define a graph convolutional operator as an aggregation of features across neighborhoods; defining
SplineNets
Parametrize the smooth spectral filter function
Spectral graph ConvNets with polynomial filters
Represent smooth spectral functions with polynomials of Laplacian eigenvalues $$w_{\alpha}(\lambda)={\sum}{j=0}^r{\alpha}{j} {\lambda}^j$$
where
Convolutional layer: Apply spectral filter to feature signal
Such graph convolutional layers are GPU friendly.
ChebNet
Graph convolution network always deal with unstructured data sets where the graph has different size. What is more, the graph is dynamic, and we need to apply to new nodes without model retraining.
Graph convolution with (non-orthogonal) monomial basis
Kipf and Welling proposed the ChebNet (arXiv:1609.02907) to approximate the filter using Chebyshev polynomial.
Application of the filter with the scaled Laplacian:
$$w_{\alpha}(\tilde{\mathbf{\Delta}})f= {\sum}{j=0}^r{\alpha}{j} T_j({\tilde{\mathbf{\Delta}}}) f={\sum}{j=0}^r{\alpha}{j}X^{(j)}$$
with
- Graph Convolutional Neural Network (Part I)
- https://www.ntu.edu.sg/home/xbresson/
- https://github.com/xbresson
Simplified ChebNets
Use Chebychev polynomials of degree
Further constrain
PinSage


In the previous post, the convolution of the graph Laplacian is defined in its graph Fourier space as outlined in the paper of Bruna et. al. (arXiv:1312.6203). However, the eigenmodes of the graph Laplacian are not ideal because it makes the bases to be graph-dependent. A lot of works were done in order to solve this problem, with the help of various special functions to express the filter functions. Examples include Chebyshev polynomials and Cayley transform.
Graph Convolution Networks (GCNs) generalize the operation of convolution from traditional data (images or grids) to graph data.
The key is to learn a function f to generate
a node
CayleyNet
Defining filters as polynomials applied over the eigenvalues of the graph Laplacian, it is possible
indeed to avoid any eigen-decomposition and realize convolution by means of efficient sparse routines
The main idea behind CayleyNet is to achieve some sort of spectral zoom property by means of Cayley transform:
$$
C(\lambda) = \frac{\lambda - i}{\lambda + i}
$$
Instead of Chebyshev polynomials, it approximates the filter as:
$$
g(\lambda) = c_0 + \sum_{j=1}^{r}[c_jC^{j}(h\lambda) + c_j^{\ast} C^{j^{\ast}}(h\lambda)]
$$
where ChebNet.
- CayleyNets: Graph Convolutional Neural Networks with Complex Rational Spectral Filters
- CayleyNets at IEEE
MotifNet
MotifNet is aimed to address the directed graph convolution.

- MotifNet: a motif-based Graph Convolutional Network for directed graphs
- Neural Motifs: Scene Graph Parsing with Global Context (CVPR 2018)
- GCN Part II @datawarrior
- http://mirlab.org/conference_papers/International_Conference/ICASSP%202018/pdfs/0006852.pdf
Minimal inner structures:
- Invariant by vertex re-indexing (no graph matching is required)
- Locality (only neighbors are considered) Weight sharing (convolutional operations)
- Independence w.r.t. graph size
Higher-order Graph Convolutional Networks
- Higher-order Graph Convolutional Networks
- A Higher-Order Graph Convolutional Layer
- MixHop: Higher-Order Graph Convolutional Architectures via Sparsified Neighborhood Mixing
- https://zhuanlan.zhihu.com/p/62300527
- https://zhuanlan.zhihu.com/p/64498484
- https://zhuanlan.zhihu.com/p/28170197
- Wavelets on Graphs via Spectral Graph Theory
- Spectral Networks and Locally Connected Networks on Graphs
Graph convolutional layer then computes a set of new node features, $(\vec{h}{1},\cdots, \vec{h}{n})$ , based on the input features as well as the graph structure.
Most prior work defines the kernels
In Graph Attention Networks the kernels
We inject the graph structure by only allowing node

- graph convolution network 有什么比较好的应用task? - 知乎
- Use of graph network in machine learning
- Node Classification by Graph Convolutional Network
- Semi-Supervised Classification with Graph Convolutional Networks
GCN for RecSys
PinSAGE Node’s neighborhood defines a computation graph. The key idea is to generate node embeddings based on local neighborhoods. Nodes aggregate information from their neighbors using neural networks.
- Graph Neural Networks for Social Recommendation
- 图神经网+推荐
- Graph Convolutional Neural Networks for Web-Scale Recommender Systems
- Graph Convolutional Networks for Recommender Systems
GCN for Bio & Chem
- DeepChem is a Python library democratizing deep learning for science.
- Chemi-Net: A molecular graph convolutional network for accurate drug property prediction
- Chainer Chemistry: A Library for Deep Learning in Biology and Chemistry
- Release Chainer Chemistry: A library for Deep Learning in Biology and Chemistry
- Modeling Polypharmacy Side Effects with Graph Convolutional Networks
- http://www.grakn.ai/?ref=Welcome.AI
- AlphaFold: Using AI for scientific discovery
- A graph-convolutional neural network model for the prediction of chemical reactivity
- Convolutional Networks on Graphs for Learning Molecular Fingerprints
GCN for NLP
- https://www.akbc.ws/2019/
- http://www.akbc.ws/2017/slides/ivan-titov-slides.pdf
- https://github.com/icoxfog417/graph-convolution-nlp
- https://nlp.stanford.edu/pubs/zhang2018graph.pdf
- https://cs.stanford.edu/people/jure/
- https://github.com/alibaba/euler
- https://ieeexplore.ieee.org/document/8439897
- Higher-order organization of complex networks
- Geometric Matrix Completion with Recurrent Multi-Graph Neural Networks
- http://snap.stanford.edu/proj/embeddings-www/
- http://ryanrossi.com/
- http://www.ipam.ucla.edu/programs/workshops/geometry-and-learning-from-data-tutorials/
- https://zhuanlan.zhihu.com/p/51990489
- https://www.cs.toronto.edu/~yujiali/
- Python for NLP
- Deep Learning on Graphs: A Survey
- Graph-based Neural Networks
- Geometric Deep Learning
- Deep Chem
- GRAM: Graph-based Attention Model for Healthcare Representation Learning
- https://zhuanlan.zhihu.com/p/49258190
- https://www.zhihu.com/question/54504471
- http://sungsoo.github.io/2018/02/01/geometric-deep-learning.html
- https://rusty1s.github.io/pytorch_geometric/build/html/notes/introduction.html
- .mp4 illustration
- Deep Graph Library (DGL)
- https://github.com/alibaba/euler
- https://github.com/alibaba/euler/wiki/%E8%AE%BA%E6%96%87%E5%88%97%E8%A1%A8
- https://www.groundai.com/project/graph-convolutional-networks-for-text-classification/
- https://datawarrior.wordpress.com/2018/08/08/graph-convolutional-neural-network-part-i/
- https://datawarrior.wordpress.com/2018/08/12/graph-convolutional-neural-network-part-ii/
- http://www.cs.nuim.ie/~gunes/files/Baydin-MSR-Slides-20160201.pdf
- http://colah.github.io/posts/2015-09-NN-Types-FP/
- https://www.zhihu.com/question/305395488/answer/554847680