Home
Welcome to the BGCFlow wiki!
This WIKI will guide you through BGCFlow, a systematic workflow for the analysis of biosynthetic gene clusters across large collections of genomes (pangenomes) from internal & public datasets.
A quick and easy way to use BGCFlow using bgcflow_wrapper.
- Create a conda environment and install the BGCFlow python wrapper :
# create and activate a new conda environment
conda create -n bgcflow -c conda-forge python=3.11 pip openjdk -y # also install java for metabase
conda activate bgcflow
# install `BGCFlow` wrapper
pip install bgcflow_wrapper
# make sure to use bgcflow_wrapper version >= 0.2.7
bgcflow --version- Deploy and run BGCFlow:
# Deploy and run BGCFlow
BGCFLOW_PATH="bgcflow" # change this to your desired path for BGCFlow"
bgcflow clone $BGCFLOW_PATH # clone BGCFlow to BGCFLOW_PATH
cd $BGCFLOW_PATH # move to BGCFLOW_PATH
bgcflow init # initiate BGCFlow config and examples from template
bgcflow run -n # do a dry run, remove the flag "-n" to run the example dataset- Read more about
bgcflow_wrapperfor a detailed overview of the command line interface.

The main Snakefile workflow comprises various pipelines for data selection, functional annotation, phylogenetic analysis, genome mining, and comparative genomics for Prokaryotic datasets.
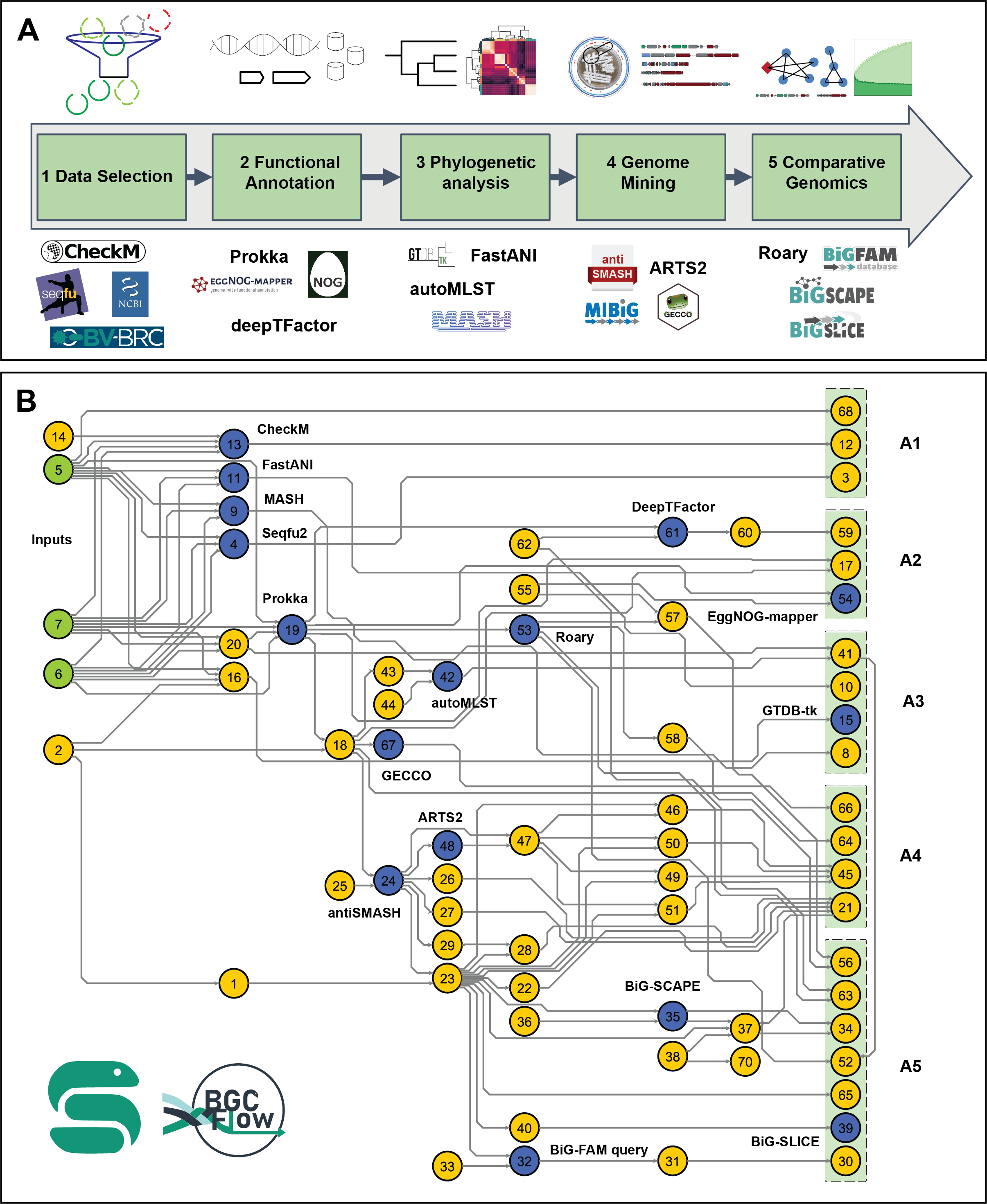
Available pipelines in the main Snakefile can be checked using the following command:
bgcflow pipelines