
| 📫 HIBLUP: Versatile and easy-to-use GS toolbox. | 🍀 SIMER: data simulation for life science and breeding. |
| 🚴♂️ KAML: Advanced GS method for complex traits. | 🏔️ IAnimal: an omics knowledgebase for animals. |
| 🏊 hibayes: A Bayesian-based GWAS and GS tool. | 📮 rMVP: Efficient and easy-to-use GWAS tool. |
CMplot is available on CRAN, so it can be installed with the following R code:
> install.packages("CMplot")
> library("CMplot")
# if you want to use the latest version on GitHub:
> source("https://raw.githubusercontent.com/YinLiLin/CMplot/master/R/CMplot.r")There are two example datasets attached in CMplot, users can export and view the details by following R code:
> data(pig60K) #calculated p-values by MLM
> data(cattle50K) #calculated SNP effects by rrblup
> head(pig60K)
SNP Chromosome Position trait1 trait2 trait3
1 ALGA0000009 1 52297 0.7738187 0.51194318 0.51194318
2 ALGA0000014 1 79763 0.7738187 0.51194318 0.51194318
3 ALGA0000021 1 209568 0.7583016 0.98405289 0.98405289
4 ALGA0000022 1 292758 0.7200305 0.48887140 0.48887140
5 ALGA0000046 1 747831 0.9736840 0.22096836 0.22096836
6 ALGA0000047 1 761957 0.9174565 0.05753712 0.05753712
> head(cattle50K)
SNP chr pos Somatic cell score Milk yield Fat percentage
1 SNP1 1 59082 0.000244361 0.000484255 0.001379210
2 SNP2 1 118164 0.000532272 0.000039800 0.000598951
3 SNP3 1 177246 0.001633058 0.000311645 0.000279427
4 SNP4 1 236328 0.001412865 0.000909370 0.001040161
5 SNP5 1 295410 0.000090700 0.002202973 0.000351394
6 SNP6 1 354493 0.000110681 0.000342628 0.000105792
As the example datasets, the first three columns are names, chromosome, position of SNPs respectively, the rest of columns are the pvalues of GWAS or effects of GS/GP for traits, the number of traits is unlimited. Note: if plotting SNP_Density, only the first three columns are needed.
Now CMplot could handle not only Genome-wide association study results, but also SNP effects, Fst, tajima's D and so on.
Total 50~ parameters are available in CMplot, typing ?CMplot can get the detail function of all parameters.
CMplot has been integrated into our developed GWAS package rMVP, please cite the following paper:
Yin, L. et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated tool for Genome-Wide Association Study, Genomics, Proteomics & Bioinformatics (2021), doi: 10.1016/j.gpb.2020.10.007.
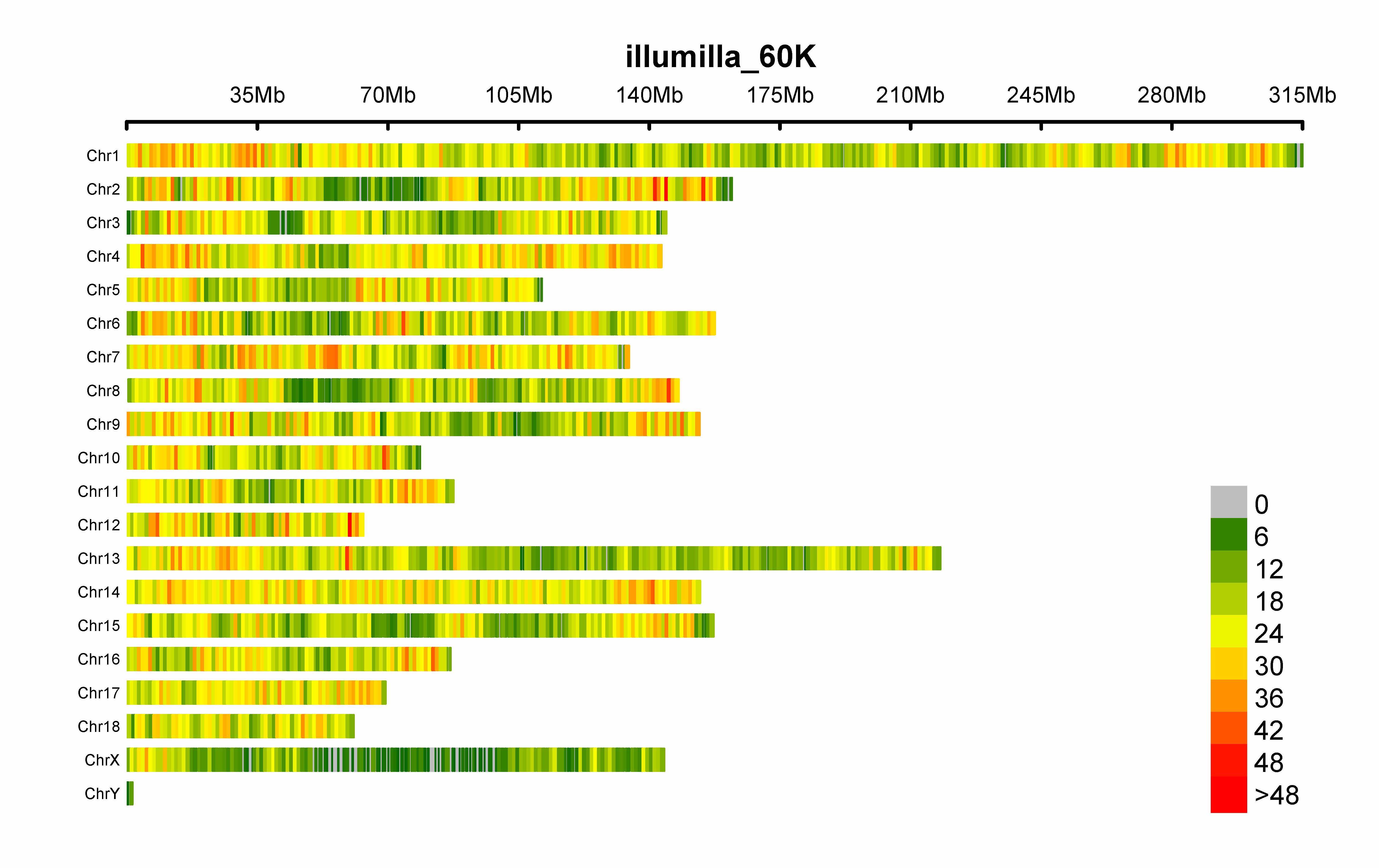
> CMplot(pig60K,plot.type="d",bin.size=1e6,chr.den.col=c("darkgreen", "yellow", "red"),file="jpg",file.name="",dpi=300,
main="illumilla_60K",file.output=TRUE,verbose=TRUE,width=9,height=6)
# set the window size: bin.size=1e6
# set the legend breaks by: bin.breaks=seq(min, max, step), e.g., bin.breaks=seq(0, 50, 10), the windows out of the breaks will be plotted in the same color as min or max.
# get the detailed information of all windows: "windinfo <- CMplot(pig60K, plot.type="d", ...)"
# file: the format of the output file, if file="png", CMplot will output a transparent background file
# file.name: specify the output file name, the default is corresponding column name when setting file.name=""
# chr.labels: change the chromosome names
# main: change the title of the plots
# NOTE: to show the full length of each chromosome, users can manually add every chromosome with one SNP, whose
# position equals to the length of corresponding chromosome, then specify the parameter: CMplot(..., chr.pos.max=TRUE).
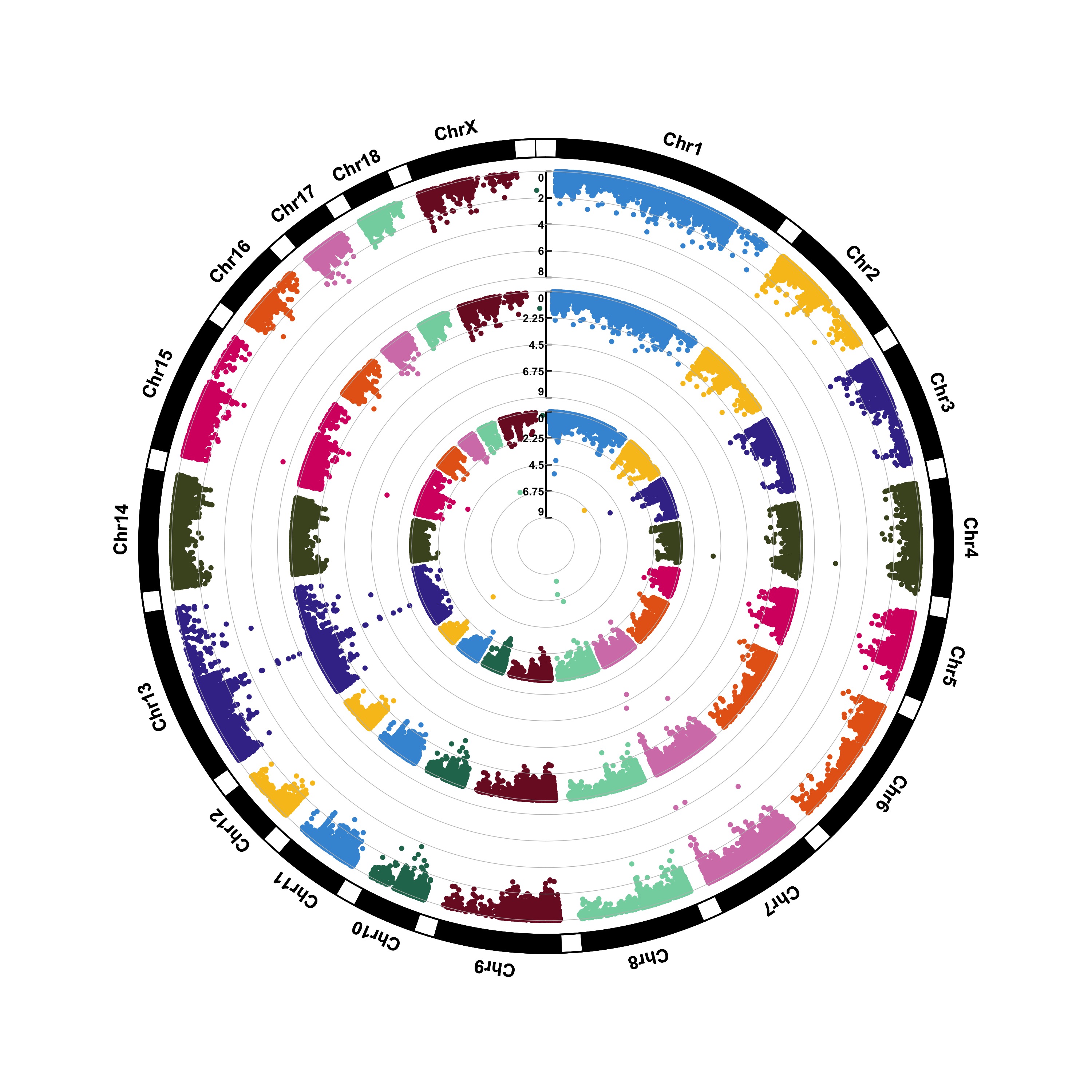
> CMplot(pig60K,type="p",plot.type="c",chr.labels=paste("Chr",c(1:18,"X","Y"),sep=""),r=0.4,cir.axis=TRUE,
outward=FALSE,cir.axis.col="black",cir.chr.h=1.3,chr.den.col="black",file="jpg",
file.name="",dpi=300,file.output=TRUE,verbose=TRUE,width=10,height=10)
# to remove the grid line in circles, add parameter cir.axis.grid=FALSE
# file.name: specify the output file name, the default is corresponding column name
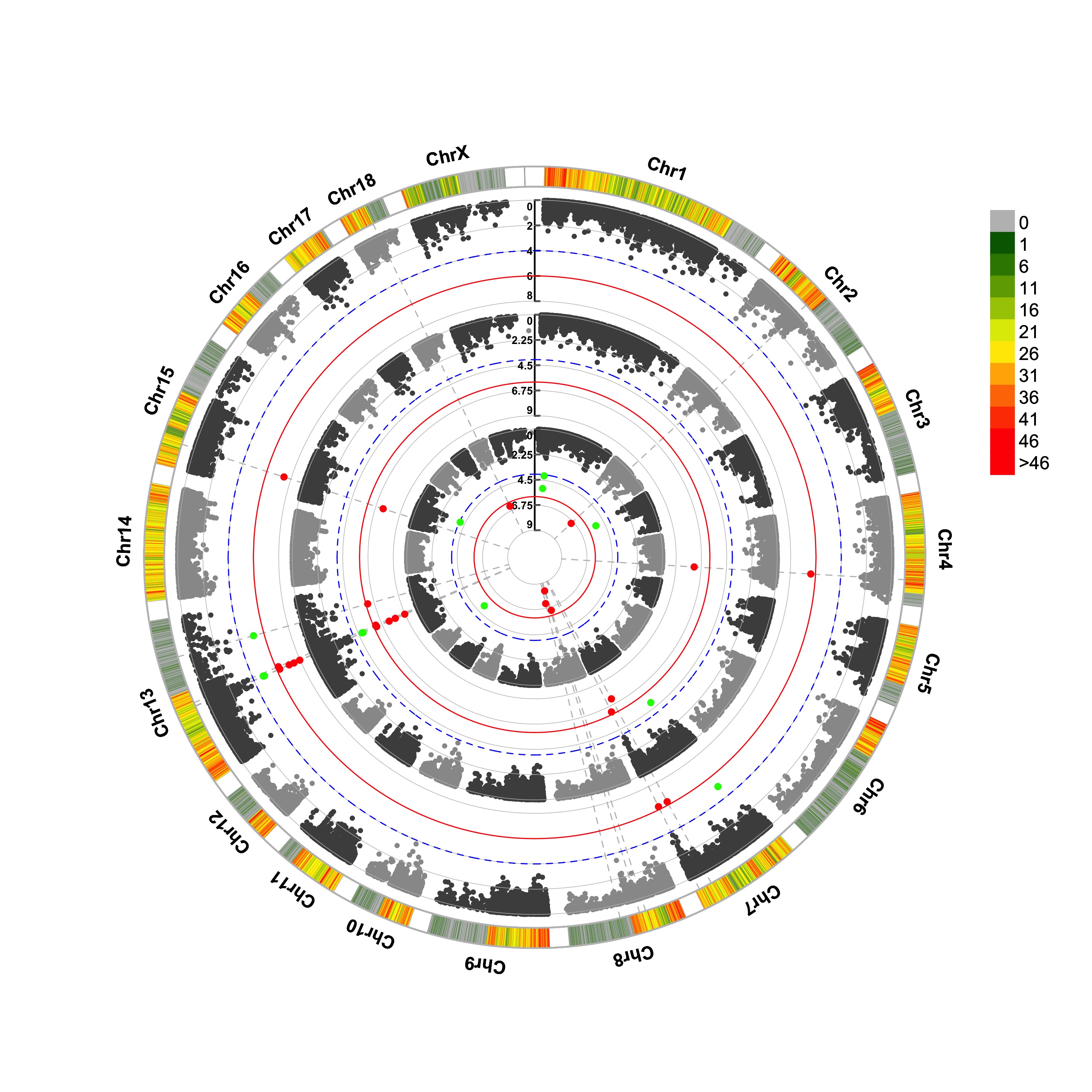
> CMplot(pig60K,type="p",plot.type="c",r=0.4,col=c("grey30","grey60"),chr.labels=paste("Chr",c(1:18,"X","Y"),sep=""),
threshold=c(1e-6,1e-4),cir.chr.h=1.5,amplify=TRUE,threshold.lty=c(1,2),threshold.col=c("red",
"blue"),signal.line=1,signal.col=c("red","green"),chr.den.col=c("darkgreen","yellow","red"),
bin.size=1e6,outward=FALSE,file="jpg",file.name="",dpi=300,file.output=TRUE,verbose=TRUE,width=10,height=10)
#Note:
1. if signal.line=NULL, the lines that crosse circles won't be added.
2. if the length of parameter 'chr.den.col' is not equal to 1, SNP density that counts
the number of SNP within given size('bin.size') will be plotted around the circle.
> CMplot(cattle50K,type="p",plot.type="c",LOG10=FALSE,outward=TRUE,col=matrix(c("#4DAF4A",NA,NA,"dodgerblue4",
"deepskyblue",NA,"dodgerblue1", "olivedrab3", "darkgoldenrod1"), nrow=3, byrow=TRUE),
chr.labels=paste("Chr",c(1:29),sep=""),threshold=NULL,r=1.2,cir.chr.h=1.5,axis.cex=1,
cir.band=1,file="jpg", file.name="",dpi=300,chr.den.col="black",file.output=TRUE,verbose=TRUE,
width=10,height=10)
# parameter 'col' can be either vector or matrix, if a matrix, each trait can be plotted in different colors.
# file.name: specify the output file name, the default is corresponding column name when setting ' file.name="" '

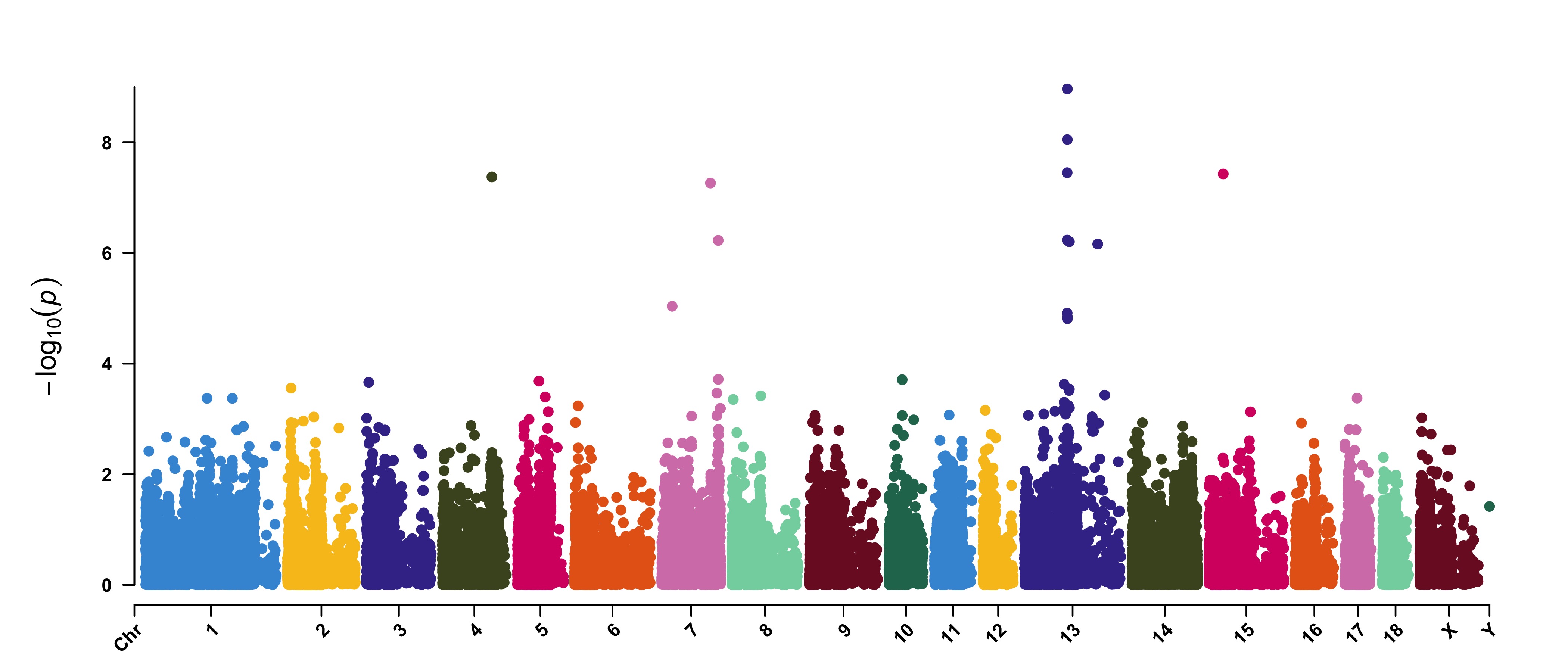
> CMplot(pig60K,type="p",plot.type="m",LOG10=TRUE,threshold=NULL,file="jpg",file.name="",dpi=300,
file.output=TRUE,verbose=TRUE,width=14,height=6,chr.labels.angle=45)
# 'chr.labels.angle': adjust the angle of labels of x-axis (-90 < chr.labels.angle < 90).
# file.name: specify the output file name, the default is corresponding column name when setting ' file.name="" '.
> CMplot(pig60K, plot.type="m", col=c("grey30","grey60"), LOG10=TRUE, ylim=c(2,12), threshold=c(1e-6,1e-4),
threshold.lty=c(1,2), threshold.lwd=c(1,1), threshold.col=c("black","grey"), amplify=TRUE,
chr.den.col=NULL, signal.col=c("red","green"), signal.cex=c(1.5,1.5),signal.pch=c(19,19),
file="jpg",file.name="",dpi=300,file.output=TRUE,verbose=TRUE,width=14,height=6)
#Note: if the ylim is setted, then CMplot will only plot the points among this interval,
# ylim can be vector or list, if it is a list, different traits can be assigned with
# different range at y-axis.
# 'threshold' can be set for different traits, for example: threshold=list(c(1e-6,1e-4), NULL, 1e-5),
# each list contains a vector of thresholds for each trait, NULL means no threshold for corresponding trait.

> CMplot(pig60K, plot.type="m", LOG10=TRUE, ylim=NULL, threshold=c(1e-6,1e-4),threshold.lty=c(1,2),
threshold.lwd=c(1,1), threshold.col=c("black","grey"), amplify=TRUE,bin.size=1e6,
chr.den.col=c("darkgreen", "yellow", "red"),signal.col=c("red","green"),signal.cex=c(1.5,1.5),
signal.pch=c(19,19),file="jpg",file.name="",dpi=300,file.output=TRUE,verbose=TRUE,
width=14,height=6)
# Note: if the length of parameter 'chr.den.col' is bigger than 1, SNP density that counts
# the number of SNP within given size('bin.size') will be plotted.
# file.name: specify the output file name, the default is corresponding column name when setting file.name=""
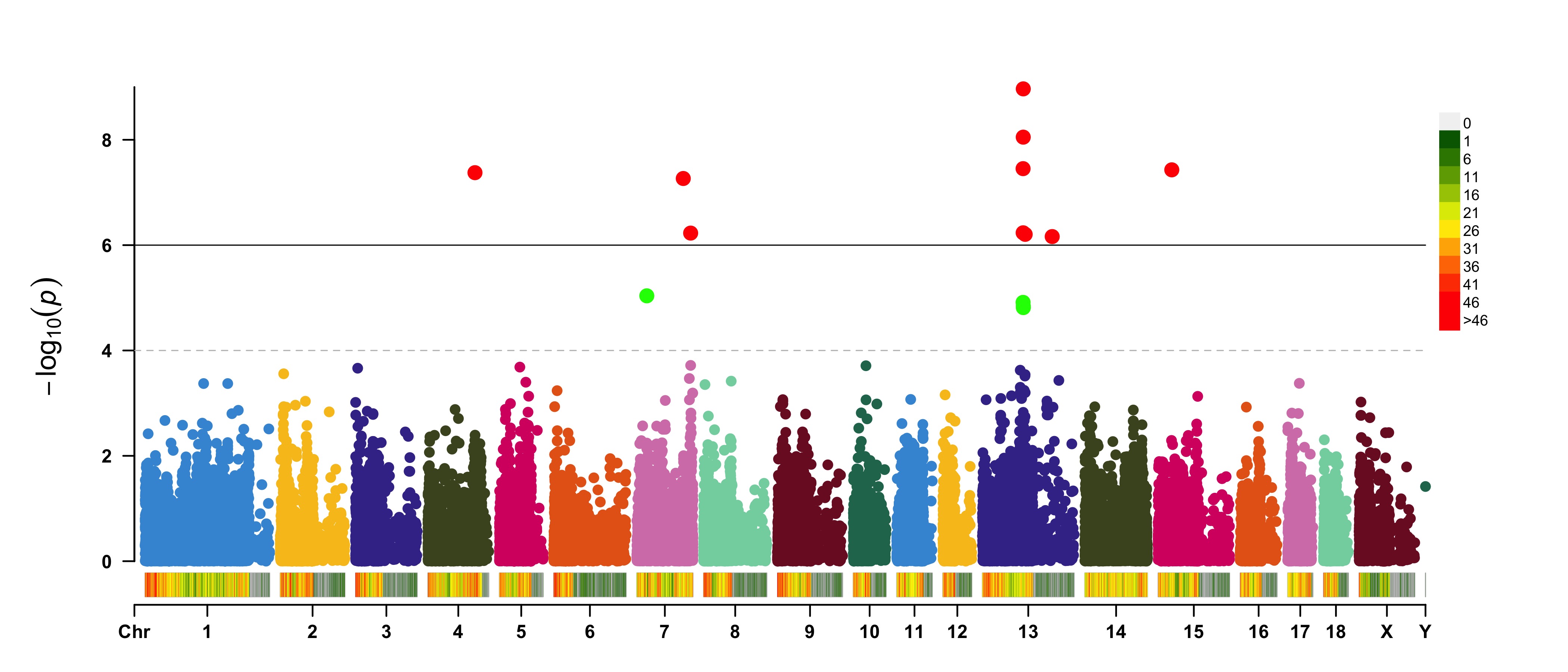
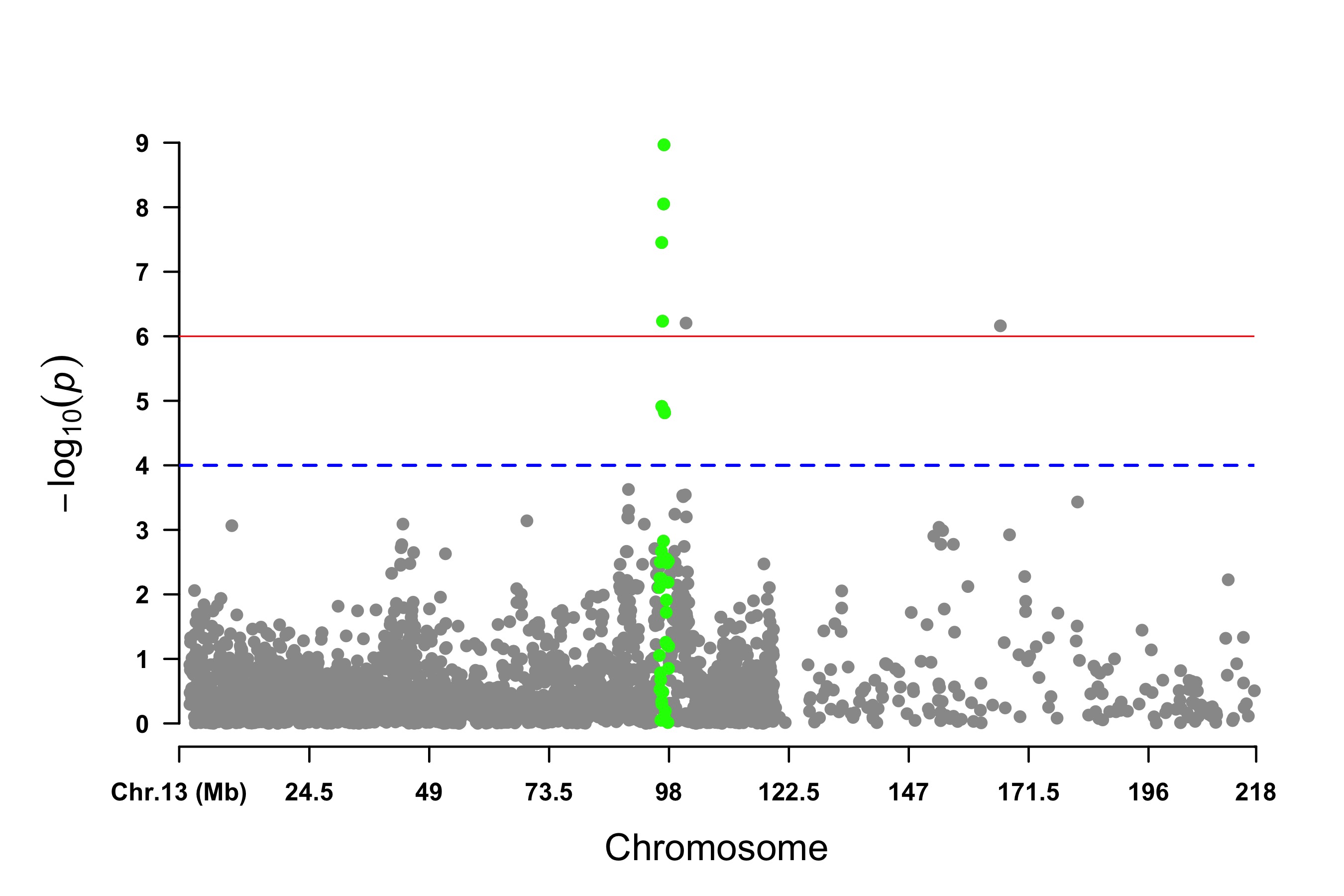
> signal <- pig60K$Position[which.min(pig60K$trait2)]
> SNPs <- pig60K$SNP[pig60K$Chromosome==13 &
pig60K$Position<(signal+1000000)&pig60K$Position>(signal-1000000)]
> CMplot(pig60K, plot.type="m",LOG10=TRUE,col=c("grey30","grey60"),highlight=SNPs,
highlight.col="green",highlight.cex=1,highlight.pch=19,file="jpg",file.name="",
chr.border=TRUE,dpi=300,file.output=TRUE,verbose=TRUE,width=14,height=6)
# Note:
# 'highlight' could be vector or list, if it is a vector, all traits will use the same highlighted SNPs index,
# if it is a list, the length of the list should equal to the number of traits.
# highlight.col, highlight.cex, highlight.pch can be value or vector, if its length equals to the length of highlighted SNPs,
# each SNPs have its special colour, size and shape.
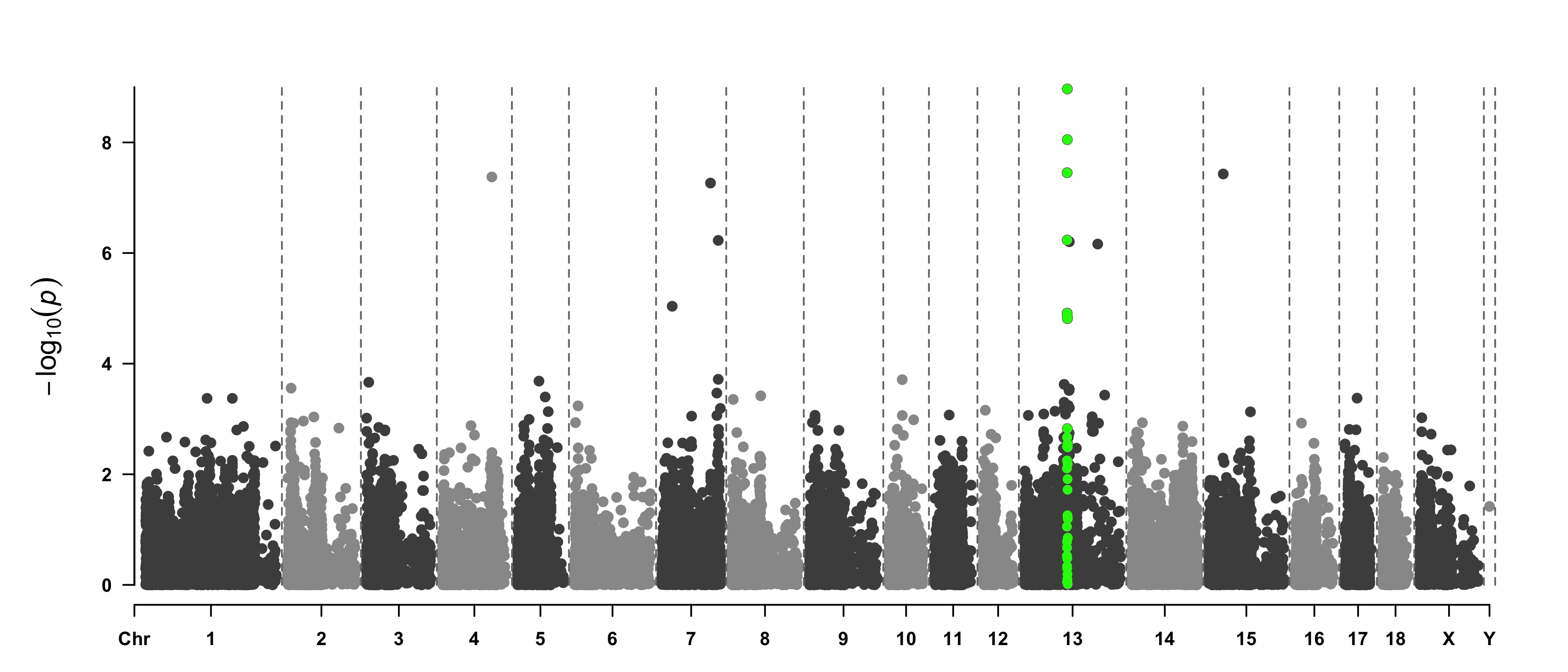
> SNPs <- pig60K[pig60K$trait2 < 1e-4, 1]
> CMplot(pig60K,type="h",plot.type="m",LOG10=TRUE,highlight=SNPs,highlight.type="p",
highlight.col=NULL,highlight.cex=1.2,highlight.pch=19,file="jpg",file.name="",
dpi=300,file.output=TRUE,verbose=TRUE,width=14,height=6,band=0.6)
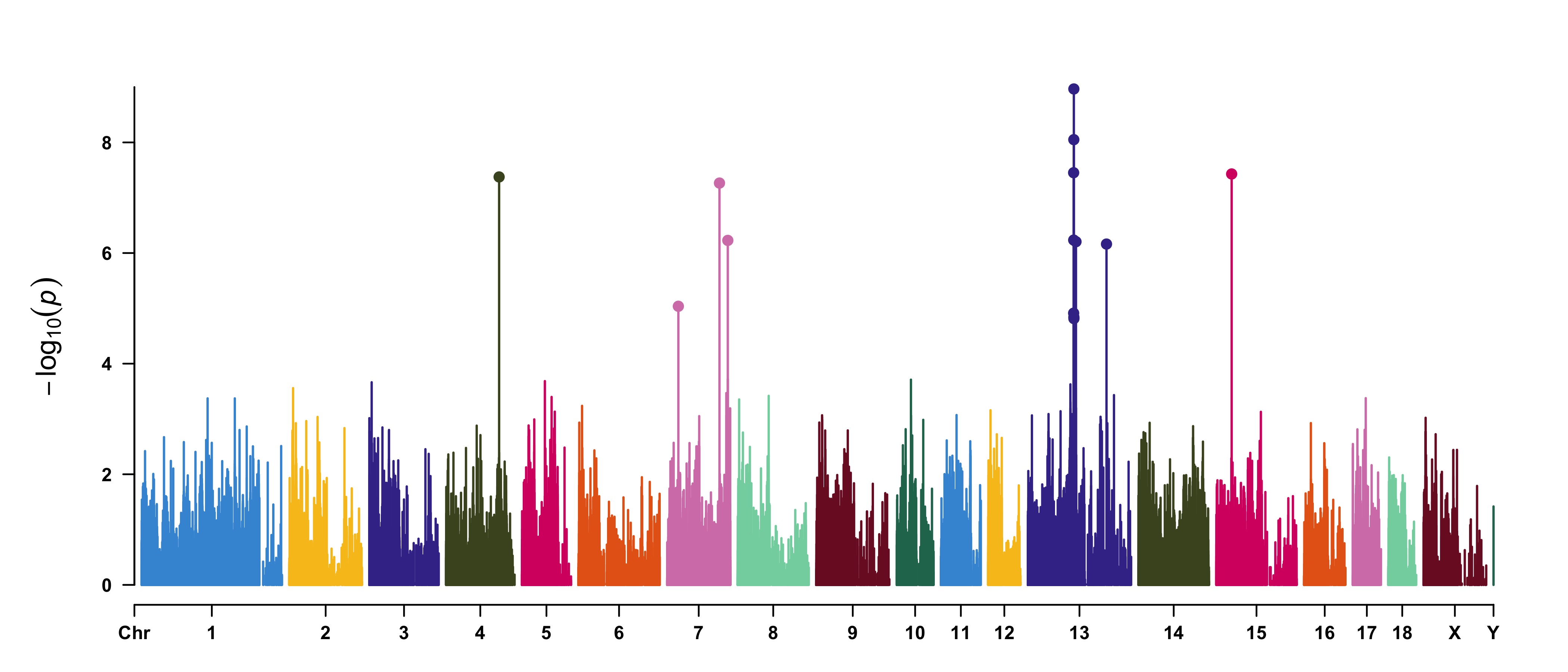
> SNPs <- pig60K[pig60K$trait2 < 1e-4, 1]
> CMplot(pig60K,type="p",plot.type="m",LOG10=TRUE,highlight=SNPs,highlight.type="h",
col=c("grey30","grey60"),highlight.col="darkgreen",highlight.cex=1.2,highlight.pch=19,
file="jpg",dpi=300,file.output=TRUE,verbose=TRUE,width=14,height=6)
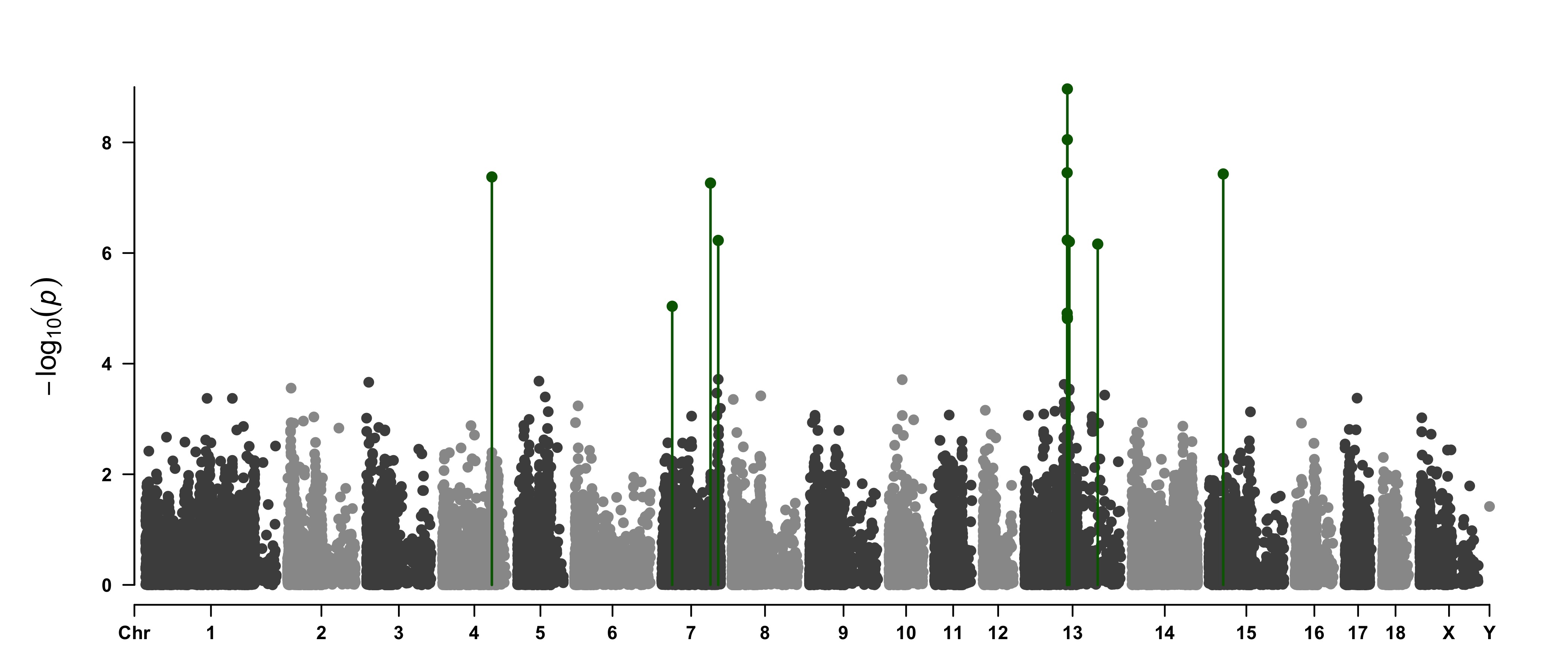
> SNPs <- pig60K[
pig60K$trait1 < 1e-4 |
pig60K$trait2 < 1e-4 |
pig60K$trait3 < 1e-4, 1]
> CMplot(pig60K,type="p",plot.type="m",LOG10=TRUE,highlight=SNPs,highlight.type="l",
threshold=1e-4,threshold.col="black",threshold.lty=1,col=c("grey60","#4197d8"),
signal.cex=1.2, signal.col="red", highlight.col="grey",highlight.cex=0.7,
file="jpg",dpi=300,file.output=TRUE,verbose=TRUE,multracks=TRUE)
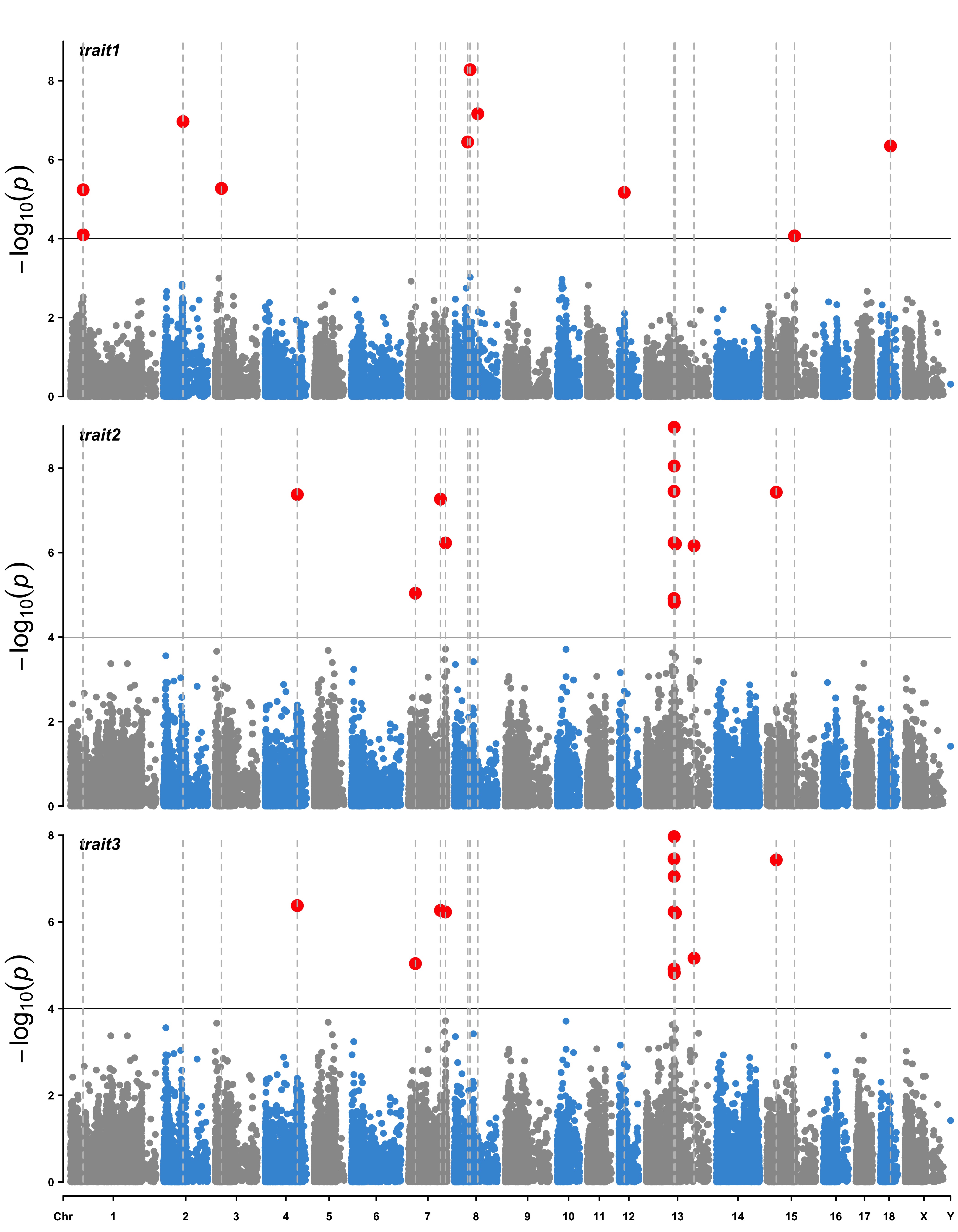
> CMplot(pig60K[pig60K$Chromosome==13, ], plot.type="m",LOG10=TRUE,col=c("grey60"),highlight=SNPs,
highlight.col="green",highlight.cex=1,highlight.pch=19,file="jpg",file.name="",
threshold=c(1e-6,1e-4),threshold.lty=c(1,2),threshold.lwd=c(1,2), width=9,height=6,
threshold.col=c("red","blue"),amplify=FALSE,dpi=300,file.output=TRUE,verbose=TRUE)
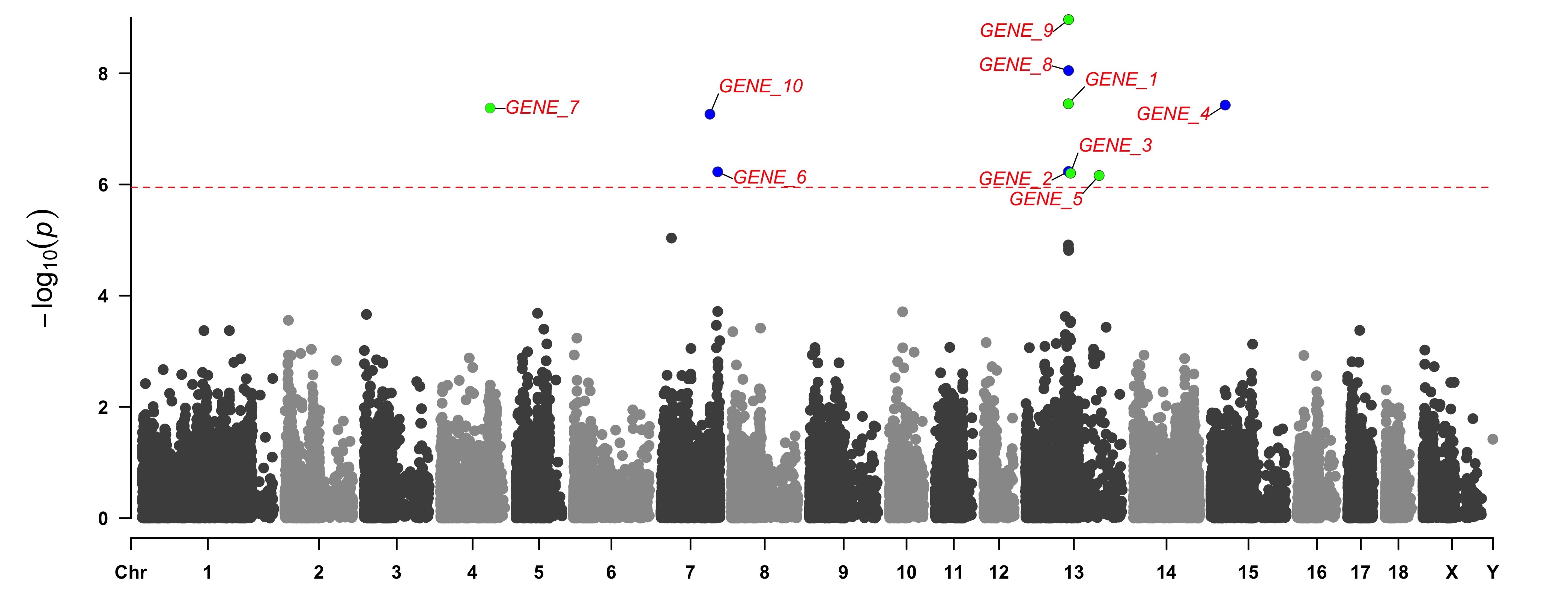
> SNPs <- pig60K[pig60K[,5] < (0.05 / nrow(pig60K)), 1]
> genes <- paste("GENE", 1:length(SNPs), sep="_")
> set.seed(666666)
> CMplot(pig60K[,c(1:3,5)], plot.type="m",LOG10=TRUE,col=c("grey30","grey60"),highlight=SNPs,
highlight.col=rep(c("green","blue"),length=length(SNPs)),highlight.cex=1, highlight.text=genes,
highlight.text.col=rep("red",length(SNPs)),threshold=0.05/nrow(pig60K),threshold.lty=2,
amplify=FALSE,file="jpg",file.name="",dpi=300,file.output=TRUE,verbose=TRUE,width=14,height=6)
# Note:
# 'highlight', 'highlight.text' could be vector or list, if it is a vector, all traits will
# use the same highlighted SNPs index and text, if it is a list, the length of the list should equal to the number of traits.
# the order of 'highlight.text' must be consistent with 'highlight'
# highlight.text.cex: value or vecter, control the size of added text
# highlight.text.font: value or vecter, control the font of added text
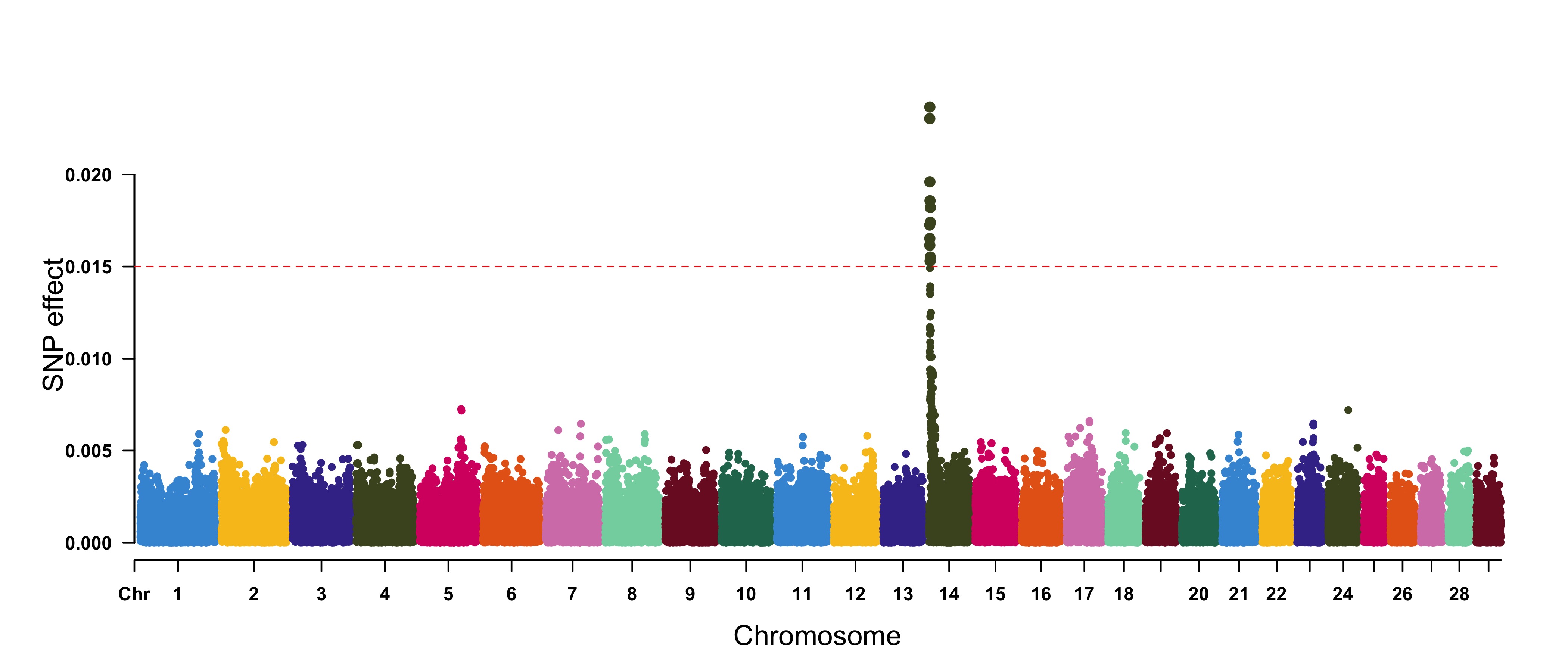
> CMplot(cattle50K, plot.type="m", band=0.5, LOG10=FALSE, ylab="SNP effect",threshold=0.015,
threshold.lty=2, threshold.lwd=1, threshold.col="red", amplify=TRUE, width=14,height=6,
signal.col=NULL, chr.den.col=NULL, file="jpg",file.name="",dpi=300,file.output=TRUE,
verbose=TRUE,cex=0.8)
#Note: if signal.col=NULL, the significant SNPs will be plotted with original colors.
> cattle50K[,4:ncol(cattle50K)] <- apply(cattle50K[,4:ncol(cattle50K)], 2,
function(x) x*sample(c(1,-1), length(x), rep=TRUE))
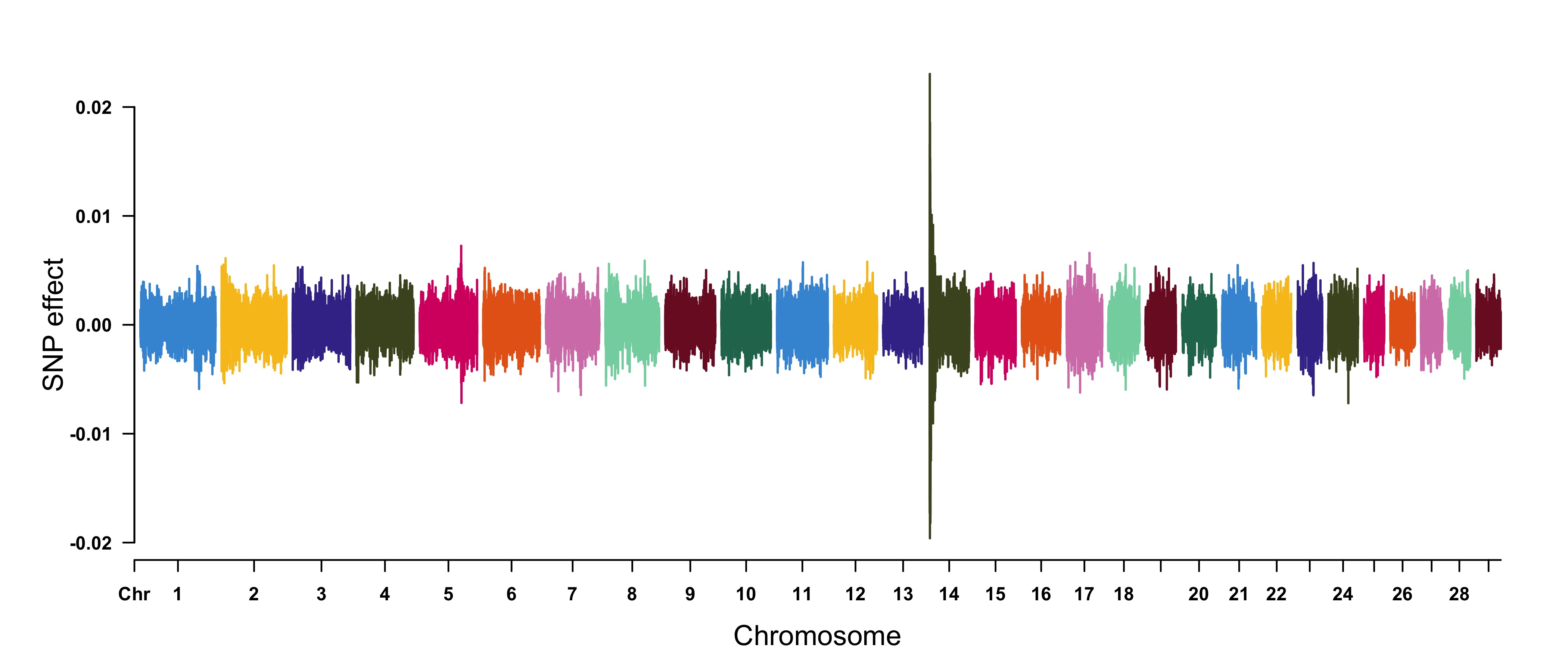
> CMplot(cattle50K, type="h",plot.type="m", band=0.5, LOG10=FALSE, ylab="SNP effect",ylim=c(-0.02,0.02),
threshold.lty=2, threshold.lwd=1, threshold.col="red", amplify=FALSE,cex=0.6,
chr.den.col=NULL, file="jpg",file.name="",dpi=300,file.output=TRUE,verbose=TRUE)
#Note: Positive and negative values are acceptable.
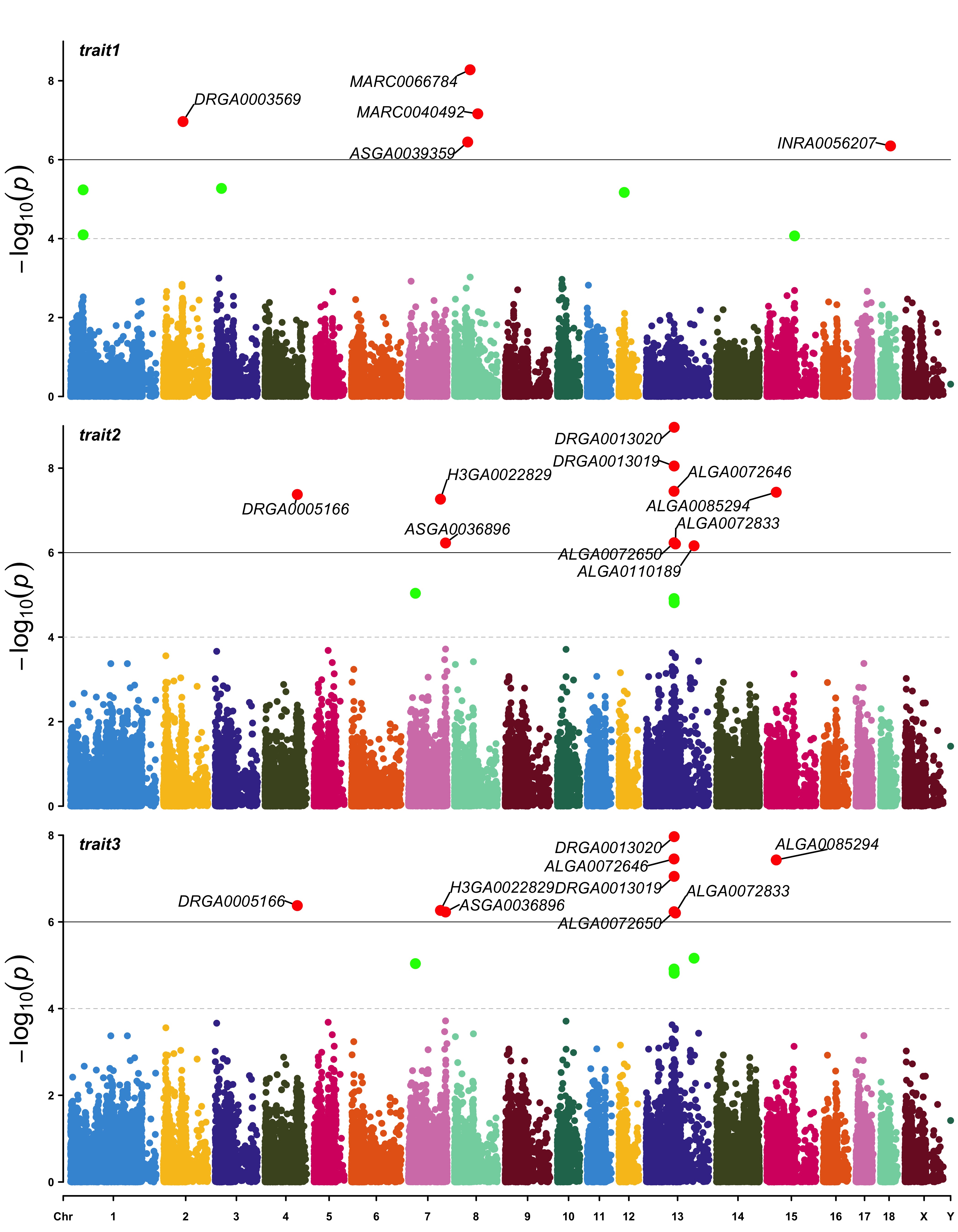
> SNPs <- list(
pig60K$SNP[pig60K$trait1<1e-6],
pig60K$SNP[pig60K$trait2<1e-6],
pig60K$SNP[pig60K$trait3<1e-6]
)
> CMplot(pig60K, plot.type="m",multracks=TRUE,threshold=c(1e-6,1e-4),threshold.lty=c(1,2),
threshold.lwd=c(1,1), threshold.col=c("black","grey"), amplify=TRUE, signal.col=
c("red","green"), signal.cex=1, file="jpg",file.name="",dpi=300,file.output=TRUE,
verbose=TRUE, highlight=SNPs, highlight.text=SNPs, highlight.text.cex=1.4)
#Note: if you are not supposed to change the color of signal,
# please set signal.col=NULL and highlight.col=NULL.
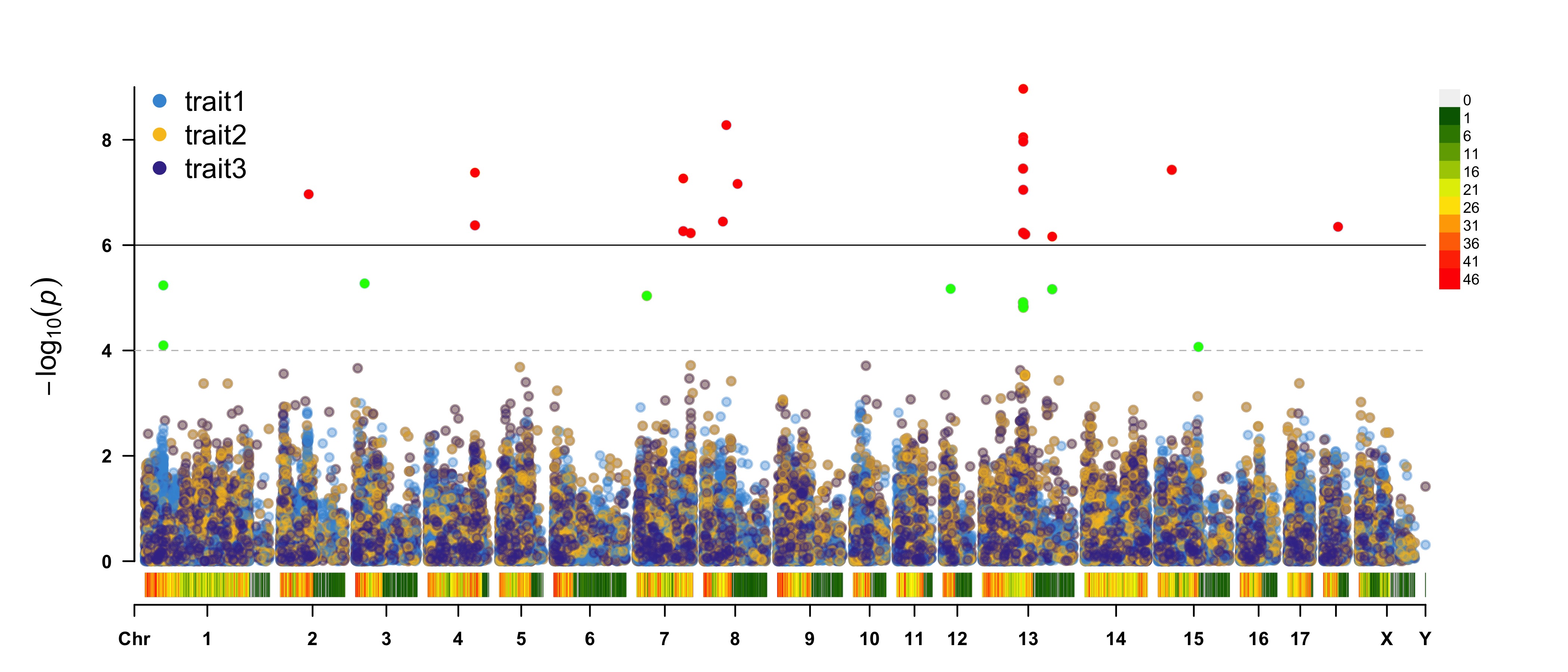
> CMplot(pig60K, plot.type="m",multraits=TRUE,threshold=c(1e-6,1e-4),threshold.lty=c(1,2),
threshold.lwd=c(1,1), threshold.col=c("black","grey"), amplify=TRUE,bin.size=1e6,
chr.den.col=c("darkgreen", "yellow", "red"), signal.col=c("red","green"),
signal.cex=1, file="jpg",file.name="",dpi=300,file.output=TRUE,verbose=TRUE,
points.alpha=100,legend.ncol=1, legend.pos="left")
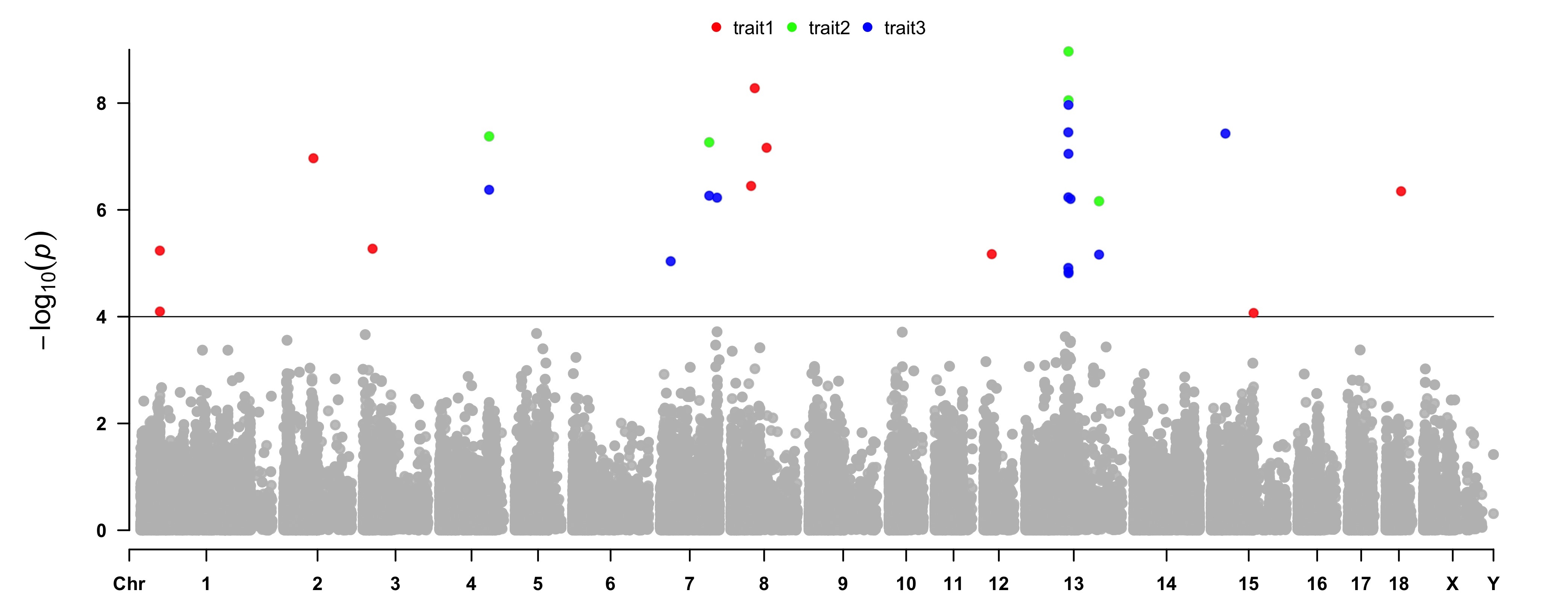
>CMplot(pig60K, plot.type="m",col="grey",multraits=TRUE,threshold=1e-4,threshold.lty=1,
threshold.lwd=c(1,1), threshold.col=c("black","grey"),amplify=TRUE,
chr.den.col=NULL, signal.col=c("red","green","blue"),signal.cex=1,
file="jpg",file.name="",dpi=300,file.output=TRUE,verbose=TRUE,
points.alpha=225,legend.ncol=3, legend.pos="middle")
# note: length of 'col' should be equal to 1 for this case.
 ---
---
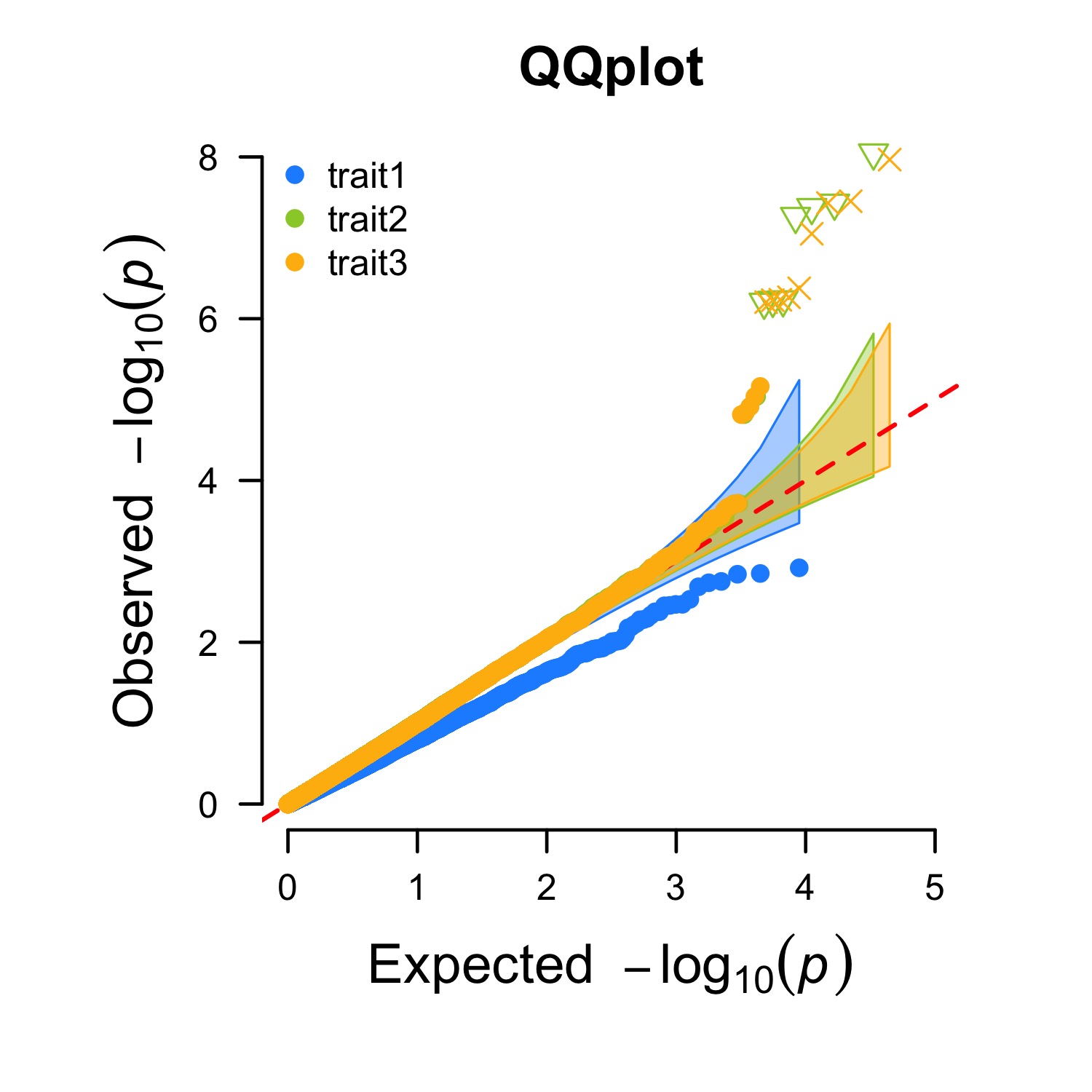
> CMplot(pig60K,plot.type="q",box=FALSE,file="jpg",file.name="",dpi=300,
conf.int=TRUE,conf.int.col=NULL,threshold.col="red",threshold.lty=2,
file.output=TRUE,verbose=TRUE,width=5,height=5)
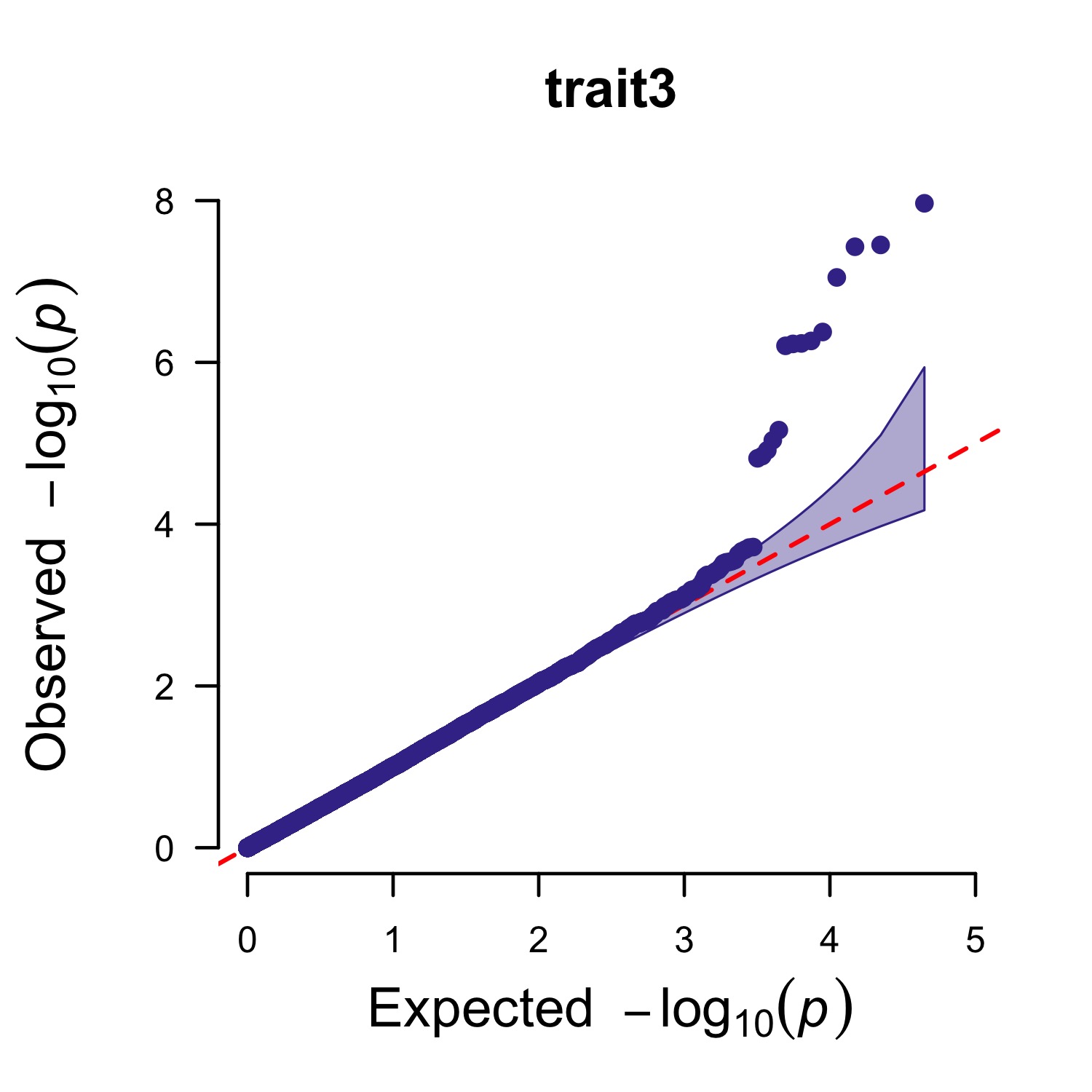
> pig60K$trait1[sample(1:nrow(pig60K), round(nrow(pig60K)*0.80))] <- NA
> pig60K$trait2[sample(1:nrow(pig60K), round(nrow(pig60K)*0.25))] <- NA
> CMplot(pig60K,plot.type="q",col=c("dodgerblue1", "olivedrab3", "darkgoldenrod1"),multracks=TRUE,
threshold=1e-6,ylab.pos=2,signal.pch=c(19,6,4),signal.cex=1.2,signal.col="red",
conf.int=TRUE,box=FALSE,axis.cex=2,file="jpg",file.name="",dpi=300,file.output=TRUE,
verbose=TRUE,ylim=c(0,8),width=5,height=5)
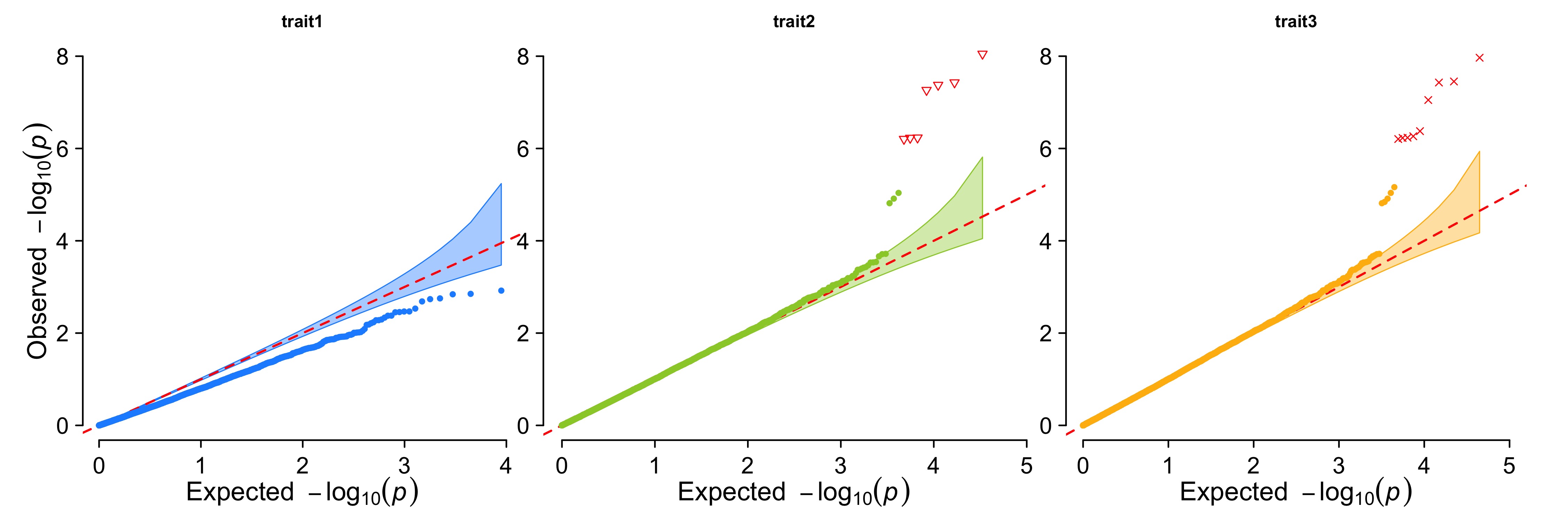
> CMplot(pig60K,plot.type="q",col=c("dodgerblue1", "olivedrab3", "darkgoldenrod1"),multraits=TRUE,
threshold=1e-6,ylab.pos=2,signal.pch=c(19,6,4),signal.cex=1.2,signal.col="red",
conf.int=TRUE,box=FALSE,axis.cex=1,file="jpg",file.name="",dpi=300,file.output=TRUE,
verbose=TRUE,ylim=c(0,8),width=5,height=5)
Questions, suggestions, and bug reports are welcome and appreciated.
- Author: Lilin Yin
- Contact: ylilin@163.com
- QQ group: 166305848
- Institution: Huazhong agricultural university