When it comes to AI for Science, arguably the most famous and successful example of AI-advanced scientific discovery is AlphaFold2 which addresses the problem of accurately predicting protein 3D structures from their sequence, one of the "holy grail" problems in structural biology. The structures of proteins are essential to their biological functions and accurate 3D modeling at the atomistic level is significant for a variety of field from drug discovery to synthetic biology. However, resolving structures experimentally is highly expensive and time-consuming, so computational methods to predict accurate protein structures have long been studied, represented by the series of CASP competitions [4]. Anfinsen's hypothesis, stating that for most proteins, the native structures in standard physiological environments are determined solely by the proteins' amino acid sequences, grounds the study of computational method for the protein structure prediction problem. Traditionally, structural biologists utilize "homology modeling" to make such predictions. In those methods, multiple proteins, whose sequences are similar to the query one and structures have already been resolved experimentally, will be found. These structures will be then be assembled to provide the predicted result. The accuracy of homology modeling depends heavily on sequence identity, and sometimes fails to reach a satisfying level. Deep learning methods, however, have stronger ability to discover correlations between sequences and structures. With carefully-designed attention-based neural networks and multiple-sequence-alignment (MSA) information, in 2020, the AlphaFold2 model achieved an astonishing average RMSD of 0.96 angstroms on the test cases in the prestigious CASP competition. Such accuracy is even comparable to experimental errors and AF2 was considered to make a breakthrough in solving this 50-year structural biological puzzle.
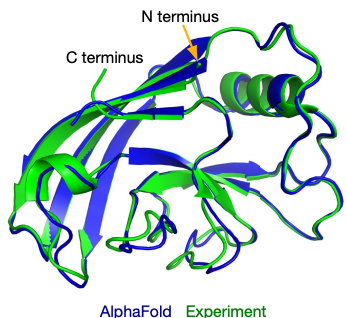
Figure 3: An illustration of AI predicted protein structure
As mentioned in the manifesto, the principles of our physical world are almost completely known but the mathematical equations are too complicated to be solved accurately. Therefore, for problems at different time and space scales, scientists have to develop different computational methods with the necessary approximations to reduce the computational complexity which is called multi-scale modeling in computational science. In this area, ab initio and classical molecular dynamics (shortened as AIMD and classical MD) are two widely used techniques. However, there has been a longstanding trade-off problem between the accuracy and efficiency of these methods. Specifically, in AIMD, the energy and forces of given systems are calculated by quantum mechanics (usually density functional theory or DFT), thus it is more accurate but more time-consuming. The computational complexity is

Figure 4: An illustration of protein-multimers