-
Notifications
You must be signed in to change notification settings - Fork 3
/
analysis_plan.Rmd
366 lines (251 loc) · 11.8 KB
/
analysis_plan.Rmd
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
330
331
332
333
334
335
336
337
338
339
340
341
342
343
344
345
346
347
348
349
350
351
352
353
354
355
356
357
358
359
360
361
362
363
364
365
---
title: "QGT-Columbia-analysis-plan"
author: "Hae Kyung Im"
date: "2020-06-03"
output: workflowr::wflow_html
editor_options:
chunk_output_type: console
---
## Set up
This information is also on the slides
- [ ] download data and software [from Box](https://uchicago.box.com/s/zhapf2zfxcpj7thvq4sjnqale3emleum).
This will have copies of all the software repositories and the models
Linux is the operating system of choice to run bioinformatics software. Here are offering two options
- Option 1: full setup, recommended for the linux-savvy with full setup
- OPtion 2: pre-installed RStudio in Google cloud, recommended for people less familiar with linux
The latest version of the analysis plan markdown document that generated this page is on [github here](https://github.com/hakyimlab/QGT-Columbia-HKI/blob/master/analysis/analysis_plan.Rmd)
rendered [here as an html page](https://hakyimlab.github.io/QGT-Columbia-HKI/analysis_plan.html)
# Option 1
- [ ] install anaconda/miniconda
- [ ] define imlabtools conda environment [how to here](https://github.com/hakyimlab/MetaXcan/blob/master/README.md#example-conda-environment-setup), which will install all the python modules needed for this analysis session
- [x] download software (copies of the repos are already included in the course folder QCT-Columbia-HKI/repos/)
- download metaxcan repo
- download torus repo
- download fastenloc repo
- download TMWR repo
- [x] download prediction models from predictdb.org (a few models are included in the course folder QCT-Columbia-HKI/repos/)
- [ ] install R/RStudio/tidyverse package
- [ ] (optional) install workflowr package in R
- [ ] git clone https://github.com/hakyimlab/QGT-Columbia-HKI.git
- [ ] start Rstudio (if you installed workflowr, you can just open the QGT-Columbia-HKI.Rproj)
# Option 2
- [ ] claim your Rstudio server IP address ()
- [ ] connect to the Rstudio server using the url you claimed (http://xxx.xxx.xxx.xxx:8787)
# Both options
- [ ] update the analysis document
```{bash eval=FALSE}
PRE="/home/student/"
cd $PRE/../lab/
git pull
```
- [ ] activate the the imlabtools environment
```{bash, eval=FALSE}
conda activate imlabtools
```
** Notice that the bash chunks need to be copy-pasted to the terminal, not performed within the chunk.
## Summary of analysis plan
- predict whole blood expression
- check how well the prediction works with GEUVADIS expression data
- run association between predicted expression and a simulated phenotype
- calculate association between expression levels and coronary artery disease risk using s-predixcan
- fine-map the coronary artery disease gwas results using torus (need some preformatting)
- calculate colocalization probability using fastenloc
- run transcriptome-wide mendelian randomization in one locus of interest
```{r preliminary definitions}
suppressPackageStartupMessages(library(tidyverse))
```
# Transcriptome-wide association methods
```{r preliminaries}
print(getwd())
##PRE="~/Box/LargeFiles/QGT-Columbia-HKI"
PRE="/home/student/QGT-Columbia-HKI"
MODEL=glue::glue("{PRE}/models")
DATA=glue::glue("{PRE}/data")
RESULTS=glue::glue("{PRE}/results")
METAXCAN=glue::glue("{PRE}/repos/MetaXcan-master/software")
FASTENLOC=glue::glue("{PRE}/repos/fastenloc-master")
TORUS=glue::glue("{PRE}/repos/torus-master")
TWMR=glue::glue("{PRE}/repos/TWMR-master")
```
```{bash folder name variables}
export PRE="/home/student/QGT-Columbia-HKI"
export DATA=$PRE/data
export MODEL=$PRE/models
export RESULTS=$PRE/results
export METAXCAN=$PRE/repos/MetaXcan-master/software
```
## predict expression
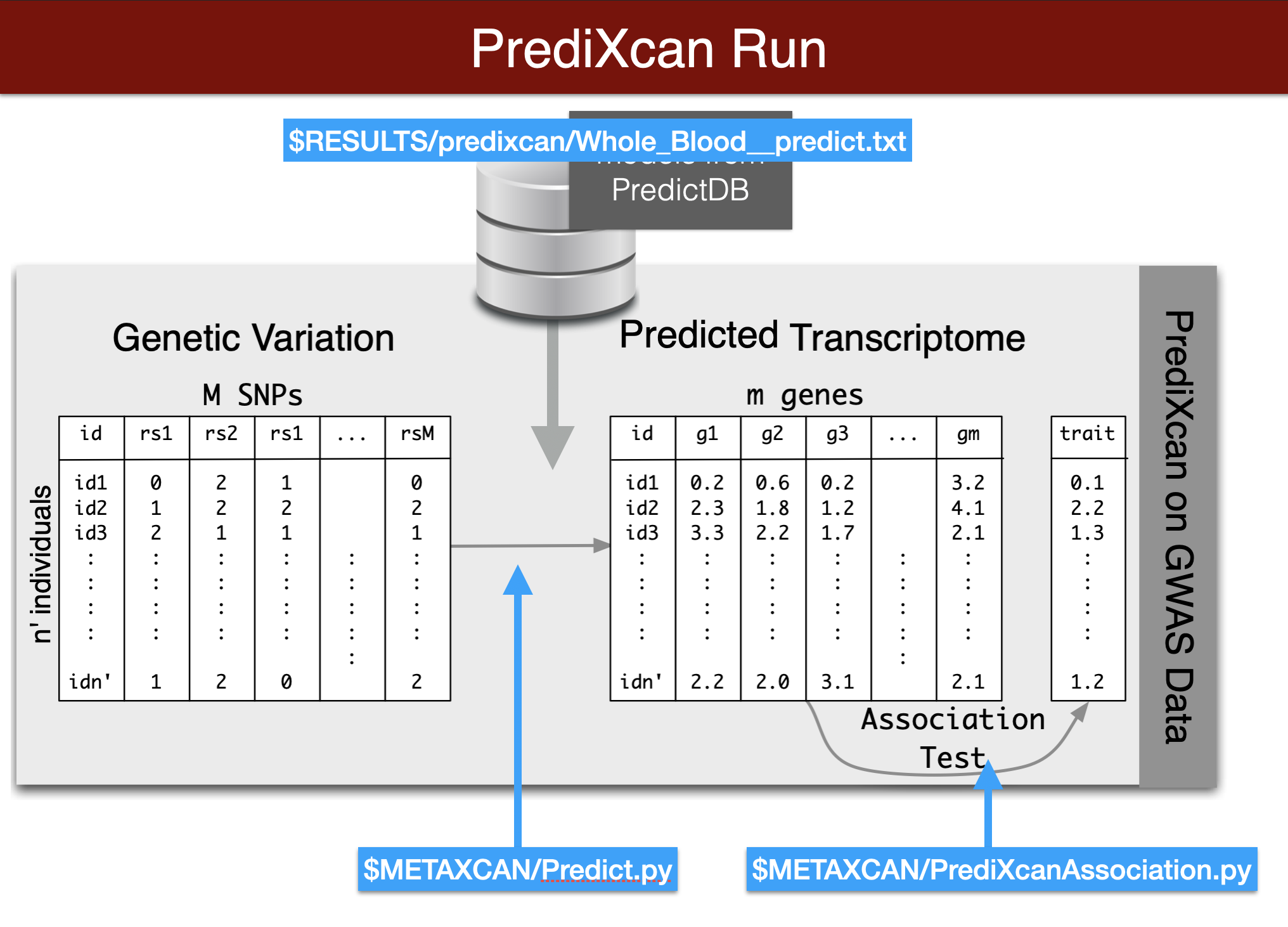
- Remember you need to copy and paste this code chunk into the terminal to run it. Also make sure you activated the imlabtools environment which has all the necessary python modules.
- Make sure all the paths and file names are correct. This run should take about one minute.
```{bash predict genetic component of expression, eval=FALSE}
printf "Predict expression\n\n"
python3 $METAXCAN/Predict.py \
--model_db_path $PRE/models/gtex_v8_en/en_Whole_Blood.db \
--vcf_genotypes $DATA/predixcan/genotype/filtered.vcf.gz \
--vcf_mode genotyped \
--variant_mapping $DATA/predixcan/gtex_v8_eur_filtered_maf0.01_monoallelic_variants.txt.gz id rsid \
--on_the_fly_mapping METADATA "chr{}_{}_{}_{}_b38" \
--prediction_output $RESULTS/predixcan/Whole_Blood__predict.txt \
--prediction_summary_output $RESULTS/predixcan/Whole_Blood__summary.txt \
--verbosity 9 \
--throw
```
## assess prediction performance (optional)
```{r assess prediction performance, eval=FALSE}
predicted_expression = read_tsv(glue::glue("{results.dir}/predixcan/Whole_Blood__predict.txt"))
dim(predicted_expression)
head(predicted_expression[,1:5])
prediction_summary = read_tsv(glue::glue("{results.dir}/predixcan/Whole_Blood__summary.txt"))
dim(prediction_summary)
head(prediction_summary)
## merge with GEUVADIS expression data
## calculate Spearman correlation
## select a few genes and plot predicted vs observed expression
```
## run association with a phenotype
```{bash run predixcan association, eval=FALSE}
PHENO="sim.infinitesimal_pve0.1"
printf "association\n\n"
python3 $METAXCAN/PrediXcanAssociation.py \
--expression_file $RESULTS/predixcan/Whole_Blood__predict.txt \
--input_phenos_file $DATA/predixcan/phenotype/$PHENO.txt \
--input_phenos_column pheno \
--output $RESULTS/predixcan/$PHENO/Whole_Blood__association.txt \
--verbosity 9 \
--throw
PHENO="sim.spike_n_slab_0.01_pve0.1"
printf "association\n\n"
python3 $METAXCAN/PrediXcanAssociation.py \
--expression_file $RESULTS/predixcan/Whole_Blood__predict.txt \
--input_phenos_file $DATA/predixcan/phenotype/$PHENO.txt \
--input_phenos_column pheno \
--output $RESULTS/predixcan/$PHENO/Whole_Blood__association.txt \
--verbosity 9 \
--throw
PHENO="sim.spike_n_slab_0.1_pve0.05"
printf "association\n\n"
python3 $METAXCAN/PrediXcanAssociation.py \
--expression_file $RESULTS/predixcan/Whole_Blood__predict.txt \
--input_phenos_file $DATA/predixcan/phenotype/$PHENO.txt \
--input_phenos_column pheno \
--output $RESULTS/predixcan/$PHENO/Whole_Blood__association.txt \
--verbosity 9 \
--throw
```
## read association results
```{r read predixcan association results, eval=FALSE}
## read association results
PHENO="sim.spike_n_slab_0.01_pve0.1"
predixcan_association = read_tsv(glue::glue("{RESULTS}/predixcan/{PHENO}/Whole_Blood__association.txt"))
## take a look at the results
dim(predixcan_association)
predixcan_association %>% arrange(pvalue) %>% head
predixcan_association %>% arrange(pvalue) %>% ggplot(aes(pvalue)) + geom_histogram(bins=20)
## compare distribution against the null (uniform)
qq(predixcan_association$pvalue)
truebetas = read_tsv(glue::glue("{DATA}/predixcan/phenotype/gene-effects/{PHENO}.txt"))
predixcan_association %>% inner_join(truebetas,by=c("gene"="gene_id")) %>% ggplot(aes(effect,effect_size))+geom_point()+geom_abline()
```
## Exercise
-[ ] show top genes
-[ ] compare with true effect sizes
```{r}
suppressPackageStartupMessages(library(qqman))
```
-------
-------
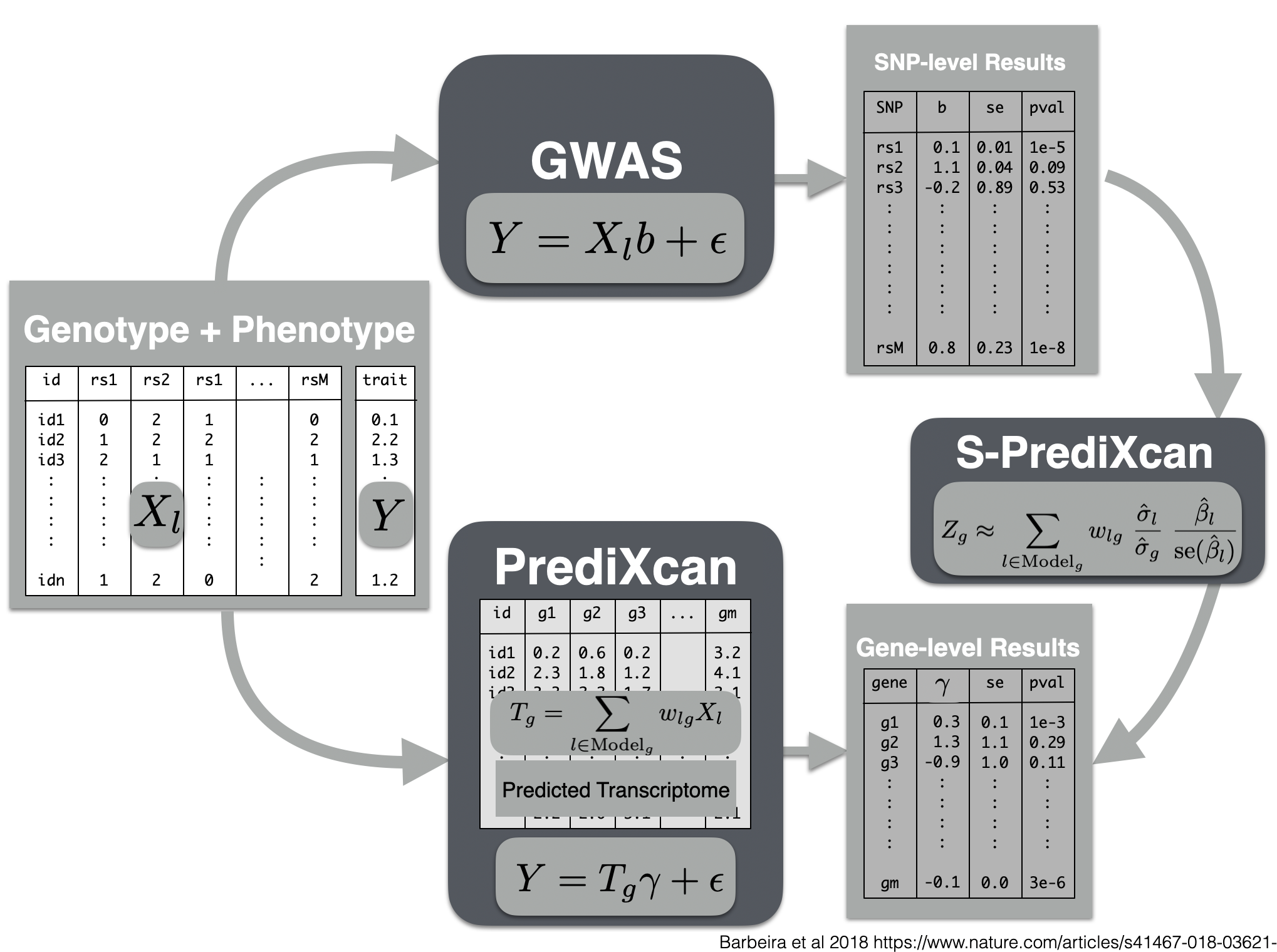
# Summary PrediXcan
Now we will use the summary results from a GWAS of coronary artery disease to calculate the association between the genetic component of the expression of genes and coronary artery disease risk. We will use the SPrediXcan.py.
```{r}
## harmonized and imputed GWAS result for coronary artery disease is available in
# $PRE/spredixcan/data/
```
## run s-predixcan
```{bash run s-predixcan, eval=FALSE}
export PRE="/home/student/QGT-Columbia-HKI"
export DATA=$PRE/data/spredixcan
export MODEL=$PRE/models
export METAXCAN=$PRE/repos/MetaXcan-master/software
export RESULTS=$PRE/results
python $METAXCAN/SPrediXcan.py \
--gwas_file $DATA/imputed_CARDIoGRAM_C4D_CAD_ADDITIVE.txt.gz \
--snp_column panel_variant_id --effect_allele_column effect_allele --non_effect_allele_column non_effect_allele --zscore_column zscore \
--model_db_path $MODEL/gtex_v8_mashr/mashr_Whole_Blood.db \
--covariance $MODEL/gtex_v8_mashr/mashr_Whole_Blood.txt.gz \
--keep_non_rsid --additional_output --model_db_snp_key varID \
--throw \
--output_file $RESULTS/spredixcan/eqtl/CARDIoGRAM_C4D_CAD_ADDITIVE__PM__Whole_Blood.csv
```
## plot and interpret s-predixcan results
```{r analyze s-predixcan results, eval=FALSE}
spredixcan_association = read_csv(glue::glue("{RESULTS}/spredixcan/eqtl/CARDIoGRAM_C4D_CAD_ADDITIVE__PM__Whole_Blood.csv"))
dim(spredixcan_association)
spredixcan_association %>% arrange(pvalue) %>% head
spredixcan_association %>% arrange(pvalue) %>% ggplot(aes(pvalue)) + geom_histogram(bins=20)
```
SORT1, considered to be a causal gene for LDL cholesterol and as a consequence of coronary artery disease, is not found here. Why? (tissue)
## run multixcan (optional)
```{bash run multixcan, eval=FALSE}
export MODEL=$PRE/models
export DATA=$PRE/data/spredixcan
python $METAXCAN/SMulTiXcan.py \
--models_folder $MODEL/gtex_v8_mashr \
--models_name_pattern "mashr_(.*).db" \
--snp_covariance $MODEL/gtex_v8_expression_mashr_snp_covariance.txt.gz \
--metaxcan_folder $RESULTS/spredixcan/eqtl/ \
--metaxcan_filter "CARDIoGRAM_C4D_CAD_ADDITIVE__PM__(.*).csv" \
--metaxcan_file_name_parse_pattern "(.*)__PM__(.*).csv" \
--gwas_file $DATA/imputed_CARDIoGRAM_C4D_CAD_ADDITIVE.txt.gz \
--snp_column panel_variant_id --effect_allele_column effect_allele --non_effect_allele_column non_effect_allele --zscore_column zscore --keep_non_rsid --model_db_snp_key varID \
--cutoff_condition_number 30 \
--verbosity 7 \
--throw \
--output $RESULTS/smultixcan/eqtl/CARDIoGRAM_C4D_CAD_ADDITIVE_smultixcan.txt
```
# Colocalization methods
- Colocalization methods seek to estimate the probability that the complex trait and expression causal variants are the same. We favor methods that calculate the probability of causality for each trait (posterior inclusion probability), called fine-mapping methods. Here we use torus for fine-mapping and fastENLOC for colocalization.
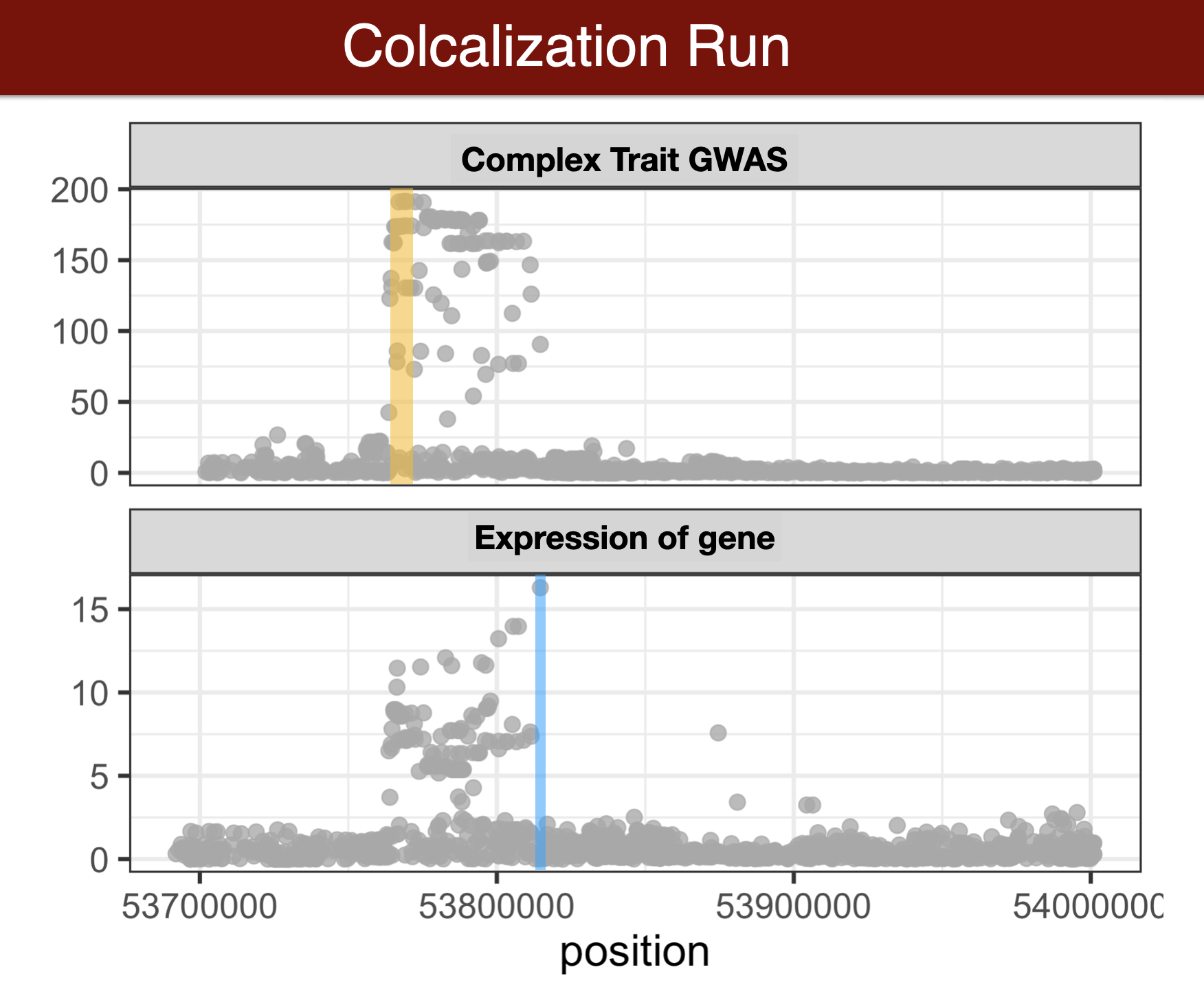
## fine-map GWAS results
We will run torus due to time limitation but ideally we would like to run a method that allows multiple causal variants per locus, such as DAP-G or SusieR.
```{bash run torus, eval=FALSE}
#torus -d Height.torus.zval.gz --load_zval -dump_pip Height.gwas.pip
#gzip Height.gwas.pip
TORUSOFT=torus
$TORUSOFT -d $PRE/data/fastenloc/Height.torus.zval.gz --load_zval -dump_pip $PRE/data/fastenloc/Height.gwas.pip
cd $PRE/data/fastenloc
gzip Height.gwas.pip
cd $PRE
```
## calculate colocalization with fastENLOC
```{bash run fastENLOC, eval=FALSE}
## check out tutorial https://github.com/xqwen/fastenloc/tree/master/tutorial
export eqtl_annotation_gzipped=$PRE/data/fastenloc/FASTENLOC-gtex_v8.eqtl_annot.vcf.gz
export gwas_data_gzipped=$PRE/data/fastenloc/Height.gwas.pip.gz
export TISSUE=Whole_Blood
export FASTENLOCSOFT=fastenloc
mkdir $RESULTS/fastenloc/
cd $RESULTS/fastenloc/
$FASTENLOCSOFT -eqtl $eqtl_annotation_gzipped -gwas $gwas_data_gzipped -t $TISSUE
#[-total_variants total_snp] [-thread n] [-prefix prefix_name] [-s shrinkage]
```
## analyze results
```{r analyze torus results}
## optional - compare with s-predixcan results
```
----------
# Mendelian randomization methods
## run TWMR (for a locus)
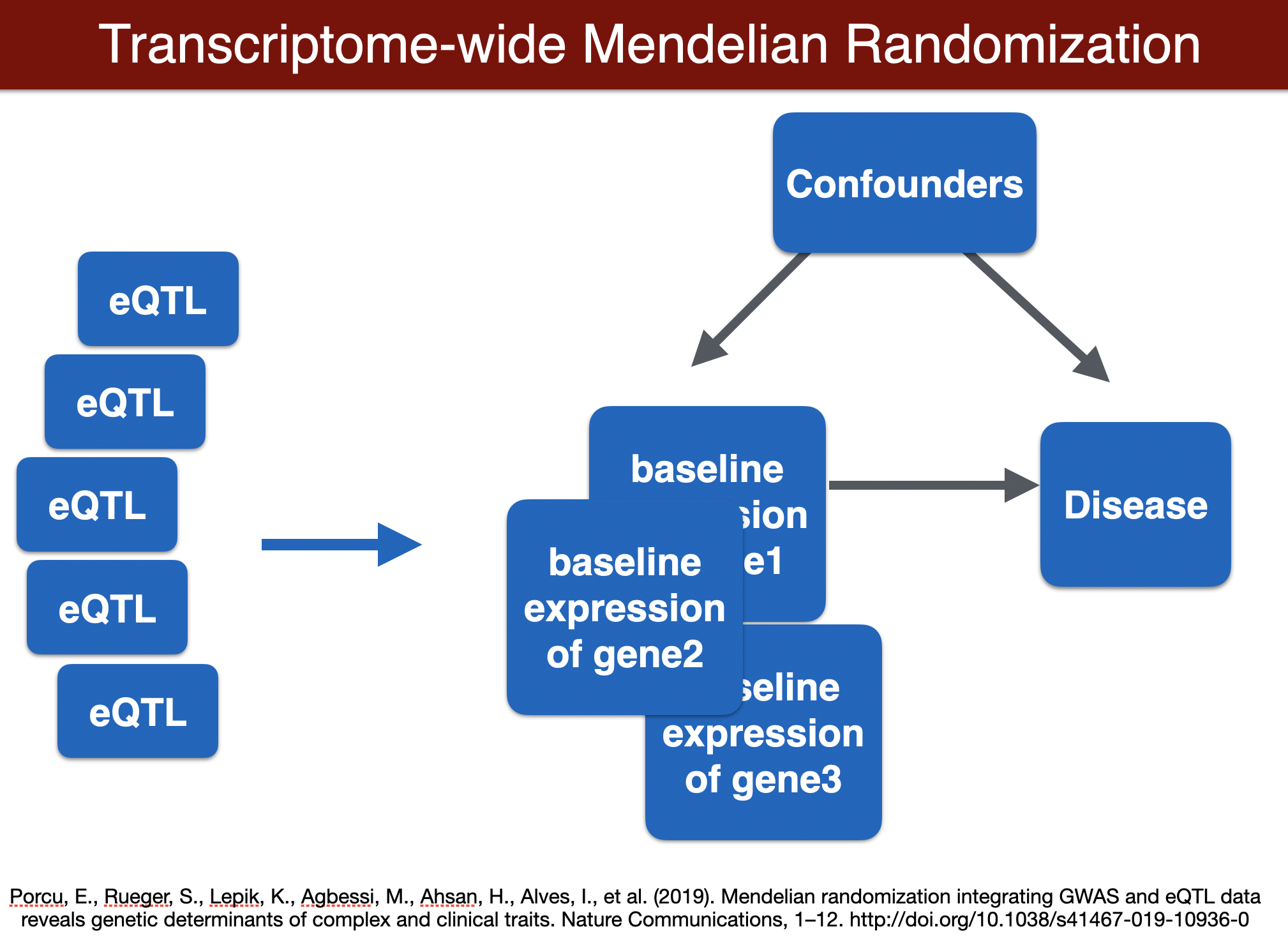
```{bash run TWMR, eval=FALSE}
export TWMR=$PRE/repos/TWMR-master
export OUTPUT=$PRE/results
GENE=ENSG00000002919
cd $TWMR
R < $TWMR/MR.R --no-save $GENE
cd $PRE
## output: /home/student/QGT-Columbia-HKI/repos/TWMR-master/ENSG00000002919.alpha
```
```{r analyze TWMR results, eval=FALSE}
```