The link to the preprint of our work: Otter-Knowledge: benchmarks of multimodal knowledge graph representation learning from different sources for drug discovery
Knowledge-enhanced learned representation enriches protein sequence and SMILES drug databases with a large multi-modal Knowledge Graph fused from different sources. This improves results on TDC drug target binding affinity prediction benchmarks.
Recent research in representation learning utilizes large databases of proteins or molecules to acquire knowledge of drug and protein structures through unsupervised learning techniques. These pre-trained representations have proven to significantly enhance the accuracy of subsequent tasks, such as predicting the affinity between drugs and target proteins. In this study, we demonstrate that by incorporating knowledge graphs from diverse sources and modalities into the sequences or SMILES representation, we can further enrich the representation and achieve state-of-the-art results on established benchmark datasets. We provide preprocessed and integrated data obtained from 7 public sources, which encompass over 30M triples. Our multimodal knowledge graphs are directed labeled graphs where each node (entities and attributes of an antity) has a modality, a particular mode that qualifies its type (text, image, numerical value, protein, molecule, etc.) and edges have labels with well-defined meanings. Additionally, we make available the pre-trained models based on this data, along with the reported outcomes of their performance on three widely-used benchmark datasets for drug-target binding affinity prediction found in the Therapeutic Data Commons (TDC) benchmarks. Additionally, we make the source code for training models on benchmark datasets publicly available. Our objective in releasing these pre-trained models, accompanied by clean data for model pretraining and benchmark results, is to encourage research in knowledge-enhanced representation learning.
We release 4 different Knowledge Graphs built from various existent datasets: Otter UBC, Otter PrimeKG, Otter DUDe and Otter STITCH.
To build these Knowledge graphs, the framework takes as input a schema file (specified in JSON) defined manually by an expert user, with good knowledge of input data sources (for example CSV or XML files, or datasets stored in a document platform such as Deep Search). The schema declaratively describes how to build the desired graph from a set of data sources. The schema defines entities (nodes in the KG) with their relevant data attributes and corresponding modality, as well as relations to other entities; these values are taken from fields present in the input data sources. Typically, one field value will be used to generate a unique identifier (UID) for the entity node, together with a namespace that indicates the origin of the entity within the original data source. Additionally, the JSON schema used as input to our framework permits user to define same_as relations among entity nodes in the graph, to represent that two nodes (potentially originating from two different input sources) are actually the same entity even if they have different UIDs. As an illustrative example of such schema, the schema used to build Otter UBC Knowledge Graph, described below, can be found in the 'schemas' folder. The framework that takes these schemas to construct the KGs will be released in the future.
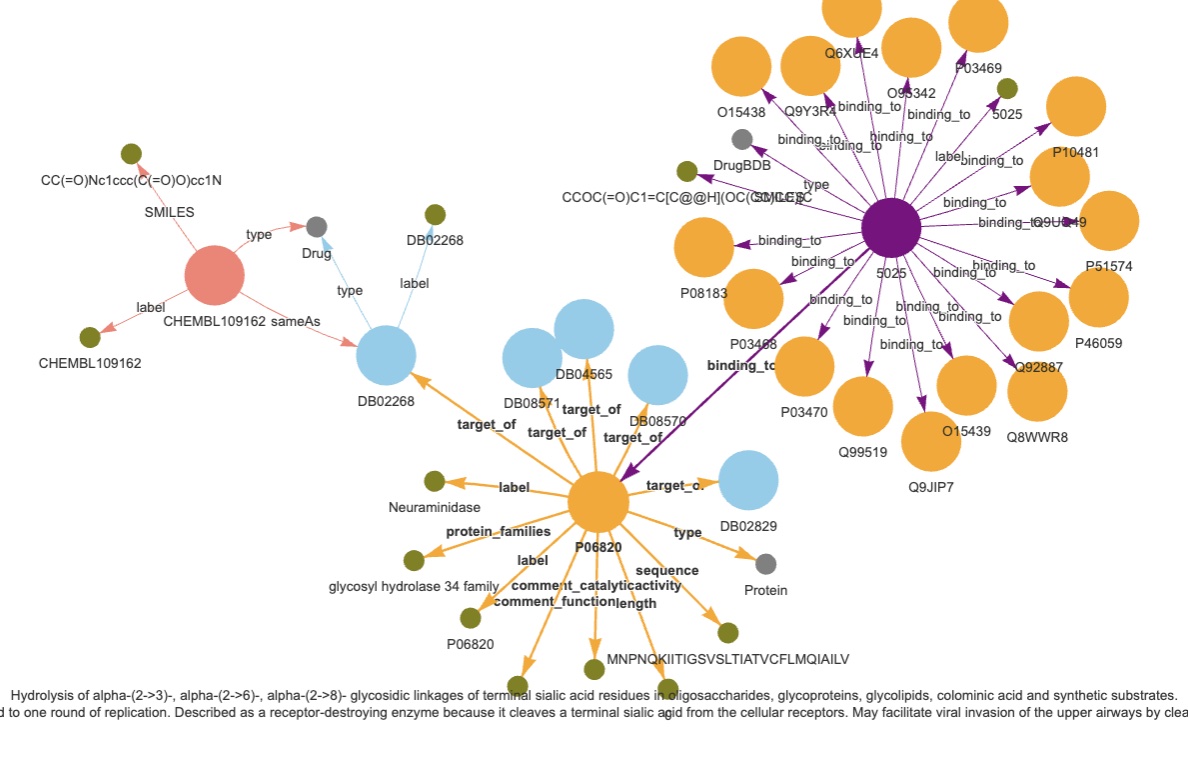
UBC is a KG dataset comprising entities (Proteins/Drugs) from Uniprot (U), BindingDB (B) and. ChemBL (C). It contains 6,207,654 triples.
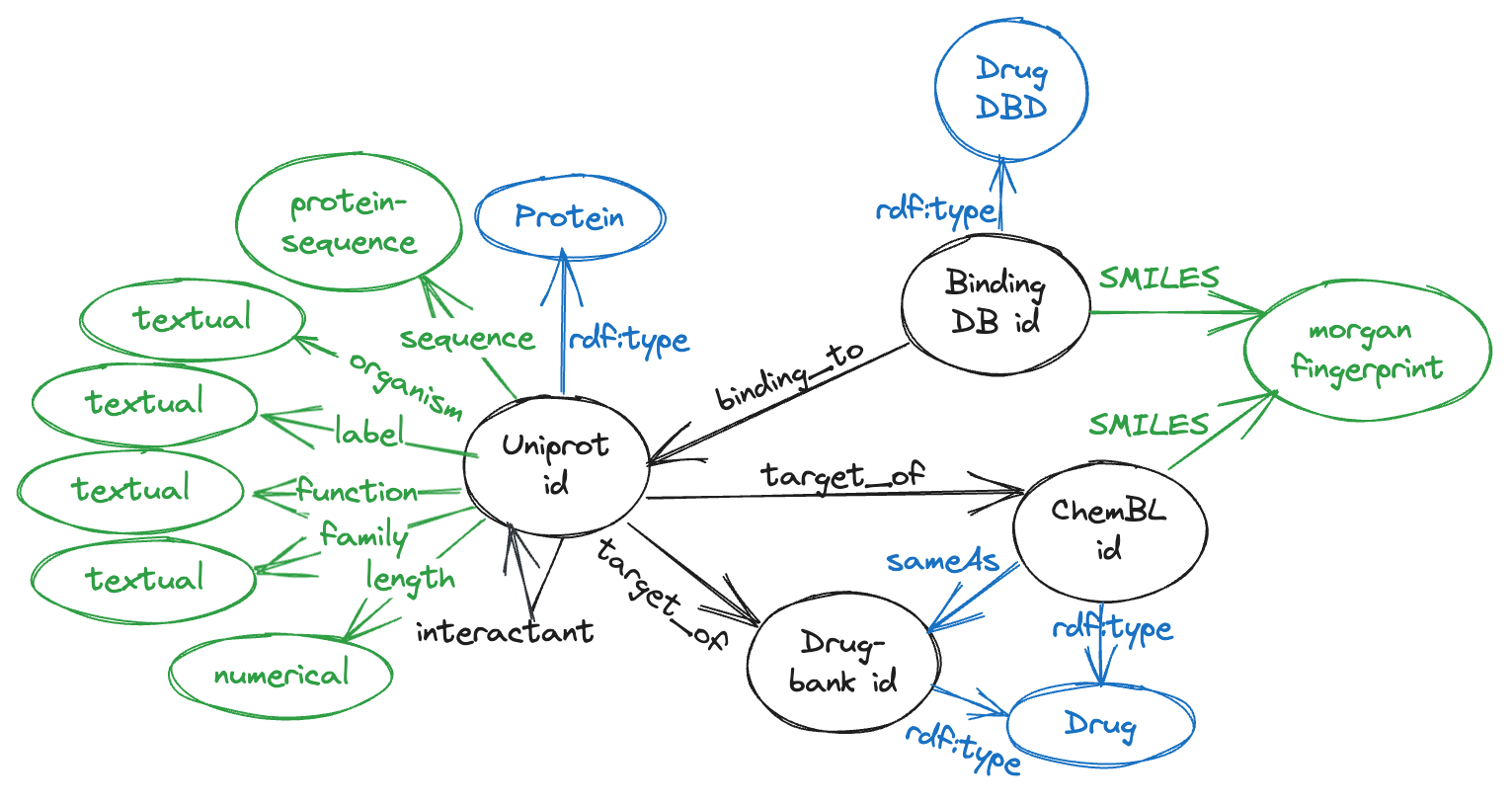
- Uniprot comprises of 573,227 proteins from SwissProt, which is the subset of manually curated entries within UniProt, including attributes with different modalities like the sequence (567,483 of them), full name, organism, protein family, description of its function, catalytics activity, pathways and its length. The number of edges are 38,665 of type target_of from Uniprot ids to both ChEMBL and Drugbank ids, and 196,133 interactants between Uniprot protein ids.
- BindingDB consists of 2,656,221 data points, involving 1.2 million compounds and 9,000 targets. Instead of utilizing the affinity score, we generate a triple for each combination of drugs and proteins. In order to prevent any data leakage, we eliminate overlapping triples with the TDC DTI dataset. As a result, the dataset concludes with a total of 2,232,392 triples.
- ChemBL comprises of drug-like bioactive molecules, 10,261 ChEMBL ids with their corresponding SMILES were downloaded from OpenTargets \cite{opentargets}, from which 7,610 have a sameAs link to drugbank id molecules.
PrimeKg (the Precision Medicine Knowledge Graph) integrates 20 biomedical resources, it describes 17,080 diseases with 4 million relationships. PrimeKG includes nodes describing Gene/Proteins (29,786) and Drugs (7,957 nodes). The MKG that we built from PrimeKG contains 13 modalities, 12,757,300 edges (154,130 data properties, and 12,603,170 object properties), including 642,150 edges describing interactions between proteins, 25,653 edges describing drug-protein interactions, and 2,672,628 describing interactions between drugs. Our multimodal KG extends the original PrimeKG graph by adding SMILES for listed drugs, and sequences for listed Uniprot proteins
DUDe comprises a collection of 22,886 active compounds and their corresponding affinities towards 102 targets. For our study, we utilized a preprocessed version of the DUDe, which includes 1,452,568 instances of drug-target interactions. To prevent any data leakage, we eliminated the negative interactions and the overlapping triples with the TDC DTI dataset. As a result, we were left with a total of 40,216 drug-target interaction pairs.
STITCH (Search Tool for Interacting Chemicals) is a database of known and predicted interactions between chemicals represented by SMILES strings and proteins whose sequences are taken from STRING database. Those interactions are obtained from computational prediction, from knowledge transfer between organisms, and from interactions aggregated from other (primary) databases. For the MKG curation we filtered only the interaction with highest confidence, i.e., the one which is higher 0.9. This resulted into 10,717,791 triples for 17,572 different chemicals and 1,886,496 different proteins. Furthermore, the graph was split into 5 roughly same size subgraphs and GNN was trained sequentially on each of them by upgrading the model trained using the previous subgraph.
Merging datasets into a single source is complex due to the difficulty of aligning their structures automatically. For example, in the STITCH knowledge graph, the connection labeled "interaction with", denoting the relationship between chemicals and proteins, is established by analyzing their co-occurrence within a Pubmed abstract. This form of association might bear resemblance to the "target of" relation found in Uniprot, however confirming their equivalence presents a challenging task because co-orcurrence in a Pubmed abstract does not mean a protein is a target of a drug. Moreover, creating a comprehensive graph by combining multiple ones demands significant computational resources.
To address these issues, we propose an approach based on ensemble methods, and we show below that it can effectively capture information from these subset of our multimodal knowledge graphs without merging them.
We release 12 models, 3 per each dataset. Otter models are based on Graph Neural Networks (GNN) that propagates initial embeddings through a set of layers that upgrade input embedding according to the node neighbours. The architecture of GNN consists of two main blocks: encoder and decoder.
- For encoder we first define a projection layer which consists of a set of linear transformations for each node modality and projects nodes into common dimensionality, then we apply several multi-relational graph convolutional layers (R-GCN) which distinguish between different types of edges between source and target nodes by having a set of trainable parameters for each edge type.
- For decoder we consider link prediction task, which consists of a scoring function that maps each triple of source and target nodes and the corresponding edge and maps that to a scalar number defined over interval [0; 1].
For link prediction, we consider three choices of scoring functions: DistMult, TransE and a Binary Classifier that are commonly used in the literature. The outcomes of scoring of each triple are then compared against actual labels using negative log likelihood loss function.
- Flow control: One crucial aspect of pretraining the GNN involves addressing the disparity between the data accessible during pretraining and the data accessible during subsequent tasks. Specifically, during pretraining, there are numerous attributes associated with proteins or drugs, whereas during downstream fine-tuning, only amino acid sequences and SMILES are available. Consequently, during pretraining, we explore two scenarios: one which controls the information propagated to the Drug/Protein entities and one without such control. In our experiments, we present results for both cases to provide an insight on the impact of restricting information flow during pretraining on the subsequent tasks.
- Noisy Links: An additional significant consideration is the presence of noisy links within the up-stream data and how they affect the downstream tasks. To investigate the potential impact on these tasks, we manually handpick a subset of links from each database that are relevant to drug discovery (see details in the Appendix). We then compare the outcomes when training the GNN using only these restricted links versus using all possible links present in the graphs.
- Regression: Certain pretraining datasets, like Uniprot, contain numerical data properties. Hence, we incorporate an extra regression objective aimed at minimizing the root mean square error (MSE) of the predicted numerical data properties. In the learning process, we combine the regression objective and the link prediction objective to create a single objective function.
| Model Name | Dataset | Scoring Type | Noisy Links | Flow Control | Regression |
|---|---|---|---|---|---|
| otter_ubc_distmult | UBC | DistMult | No | Yes | No |
| otter_ubc_transe | UBC | TransE | No | Yes | No |
| otter_ubc_classifier | UBC | Classifier Head | No | Yes | No |
| otter_primekg_distmult | PrimeKG | DistMult | No | Yes | No |
| otter_primekg_transe | PrimeKG | TransE | No | Yes | No |
| otter_primekg_classifier | PrimeKG | Classifier Head | No | Yes | No |
| otter_dude_distmult | DUDe | DistMult | No | Yes | No |
| otter_dude_transe | DUDe | TransE | No | Yes | No |
| otter_dude_classifier | DUDe | Classifier Head | No | Yes | No |
| Dataset | DTI DG | DAVIS | KIBA | ||||
|---|---|---|---|---|---|---|---|
| Splits | Temporal | Random | Target | Drug | Random | Target | Drug |
| TDC LeaderBoard | 0.538 | NA | NA | NA | NA | NA | NA |
| ESM+Morgan-Fingerprint | 0.569 | 0.805 | 0.554 | 0.264 | 0.852 | 0.630 | 0.576 |
| otter_ubc_distmult | 0.578 | 0.808 | 0.572 | 0.152 | 0.859 | 0.627 | 0.593 |
| otter_ubc_transe | 0.577 | 0.807 | 0.571 | 0.130 | 0.858 | 0.644 | 0.583 |
| otter_ubc_classifier | 0.580 | 0.810 | 0.573 | 0.104 | 0.861 | 0.631 | 0.616 |
| otter_primekg_distmult | 0.575 | 0.806 | 0.571 | 0.162 | 0.856 | 0.611 | 0.617 |
| otter_primekg_transe | 0.573 | 0.807 | 0.568 | 0.186 | 0.858 | 0.642 | 0.607 |
| otter_primekg_classifier | 0.576 | 0.813 | 0.576 | 0.133 | 0.861 | 0.630 | 0.635 |
| otter_dude_distmult | 0.577 | 0.805 | 0.573 | 0.132 | 0.857 | 0.650 | 0.607 |
| otter_dude_transe | 0.576 | 0.807 | 0.570 | 0.170 | 0.856 | 0.653 | 0.604 |
| otter_dude_classifier | 0.579 | 0.808 | 0.574 | 0.167 | 0.860 | 0.641 | 0.630 |
| otter-knowledge-ensemble | 0.588 | 0.839 | 0.578 | 0.168 | 0.886 | 0.678 | 0.638 |
Clone the repo:
git clone https://github.com/IBM/otter-knowledge.git
cd otter-knowledge
Install the requirements:
pip install -r requirements.txt
usage: inference.py [-h] --input_path INPUT_PATH [--sequence_column SEQUENCE_COLUMN] [--input_type INPUT_TYPE] [--model_path MODEL_PATH] --output_path OUTPUT_PATH [--batch_size BATCH_SIZE] [--no_cuda]
Inference
options:
-h, --help show this help message and exit
--input_path INPUT_PATH
Path to the csv file with the sequence/smiles
--sequence_column SEQUENCE_COLUMN
Name of the column with sequence/smiles information for proteins or molecules
--input_type INPUT_TYPE
Type of the sequences. Options: Drug; Protein
--model_path MODEL_PATH
Path to the model or name of the model in the HuggingfaceHub
--output_path OUTPUT_PATH
Path to the output embedding file.
--batch_size BATCH_SIZE
Batch size to use.
--no_cuda If set to True, CUDA won't be used even if available.
-
Run the inference for Proteins:
Replace test_data with the path to a CSV file containing the protein sequences, name_of_the_column with the name of the column of the protein sequence in the CSV and output_path with the filename of the JSON file to be created with the embeddings.
python inference.py --input_path test_data --sequence_column name_of_the_column --model_path ibm/otter_dude_distmult --output_path output_path-
Run the inference for Drugs:
Replace test_data with the path to a CSV file containing the Drug SMILES, name_of_the_column with the name of the column of the SMILES in the CSV and output_path with the filename of the JSON file to be created with the embeddings..*
python inference.py --input_path test_data --sequence_column name_of_the_column input_type Drug --relation_name smiles --model_path ibm/otter_dude_distmult --output_path output_pathWe assume that you have used the inference script to generate embeddings for training and test proteins/drugs. The embeddings of training and test proteins/drugs should be combined into files with the following format that keep computed embeddings of drugs/proteins. It is important to notice that the inference only generates embeddings for either drugs or proteins so you need to combine and convert them into the following format so that they can be used as input to the model benchmark training as explained below.
{
"Drug": {
"CN(C)CC(=O)NC(COc1cncc(-c2ccc3cnccc3c2)c1)Cc1c[nH]c2ccccc12": [
-1.2718517780303955,
0.6045345664024353,
-0.03671235218644142,
0.9915799498558044,
-0.7146453857421875],
"Cc1sc2ncnc(N)c2c1-c1ccc(NC(=O)Nc2cc(C(F)(F)F)ccc2F)cc1": [
-0.6596673130989075,
0.2838267683982849,
-0.042177166789770126,
0.7447476387023926,
-0.27911311388015747]
},
"Target": {
"MTLDVGPEDELPDWAAAKEFYQKYDPKDVIGRGVSSVVRRCVHRATGHE": [
-0.46595990657806396,
-0.297667533159256,
-0.048857495188713074]
}
}Training benchmark models can be done with the following example command:
python -m benchmarks.dti.train --train train_val.csv --test test.csv --train_embeddings train_val_embeddings.json --test_embeddings test_embeddings.json
Where the input to the script are:
- train_val.csv the path to the csv file that keep the training data from TDC benchmarks
- test.csv the path to the csv file that keep the test data from TDC benchmarks
- the input files train_val_embeddings.json and test_embeddings.json keeps the computed embeddings of train/test protein/drugs respectively in the format that we have discussed above.
There are other optional hyperparameter you can set such as the learning rate, the number of training steps etc as below
usage: train.py [-h] [--train TRAIN] [--test TEST] [--train_embeddings TRAIN_EMBEDDINGS] [--test_embeddings TEST_EMBEDDINGS] [--lr LR] [--steps STEPS] [--seeds SEEDS] [--batch_size BATCH_SIZE] [--is_initial_embeddings IS_INITIAL_EMBEDDINGS]
[--gnn_embedding_dim GNN_EMBEDDING_DIM]
TDC DG training
optional arguments:
-h, --help show this help message and exit
--train TRAIN Root directory with the training data
--test TEST Root directory with the test data
--train_embeddings TRAIN_EMBEDDINGS
Root directory with the embeddings of training drugs and proteins.
--test_embeddings TEST_EMBEDDINGS
Root directory with the embeddings of test drugs and proteins.
--lr LR Learning rate.
--steps STEPS Maximum number of training steps
--seeds SEEDS Random seeds.
--batch_size BATCH_SIZE
Mini batch size.
--is_initial_embeddings IS_INITIAL_EMBEDDINGS
Set this value to yes if want to train with initial embeddings without GNN embeddings.
--gnn_embedding_dim GNN_EMBEDDING_DIM
Size of the GNN embeddings.
Ensemble method combines the predictions trained on different GNN embeddings provided by different pretrained models. The following example command run ensemble learning:
python -m benchmarks.dti.train_ensemble_model --train train_val.csv --test test.csv --train_embeddings train_embeddings.txt --test_embeddings test_embeddings.txt
Where the input to the script are:
- train_val.csv the path to the csv file that keep the training data from TDC benchmarks
- test.csv the path to the csv file that keep the test data from TDC benchmarks
- the input files train_embeddings.txt and test_embeddings.txt keeps keep a list of train/test embedding files (each line in the file is the path to the computed embeddings files).
For example, the content of the train_embeddings.txt may look like follows:
train_val_embeddings_1.json
train_val_embeddings_2.json
train_val_embeddings_3.json
And the content of the test_embeddings.txt may look like follows:
test_embeddings_1.json
test_embeddings_2.json
test_embeddings_3.json
Where the train_val_embeddings_1.json and test_embeddings_1.json are the computed GNN embeddings of train/test drugs/proteins respectively using a pretrained models.