The purpose of ASSEMBLY is to merge reads into larger contiguous sequences (contigs) based on their sequence similarity to each other. The purpose of ALIGNMENT is to compare the contig to a previously assembled reference genome (or transcriptome) sequence.
Mapping
- A mapping is a region where a read sequence is placed.
- A mapping is regarded to be correct if it overlaps the true region.
Alignment
- An alignment is the detailed placement of each base in a read.
- An alignment is regarded to be correct if each base is placed correctly.
Tools used to be separated into aligners vs mappers. However these have become combined over time. However a distinction still exists. For example, studies examining SNPs and variations in a genome would be primarily alignment-oriented. However, studies focusing on RNA-Seq would be essentially mapping-oriented. The reason for this difference is that in RNA-Seq you would need to know where the measurements come from, but you would be less concerned about their proper alignment.
The limitations of alignment are:
- sequencing errors
- genomic variation
- repetitive elements
- multiple different transcript isoforms from same gene
- main challenge is the spliced alignment of exon-exon spanning reads
- mapping ambiguity = many reads overlap with more than one isoform
- False positives: lowly expressed isoforms are excluded by alignment algorithms —> bias towards identifying strongly expressed genes

- align reads to genome index to identify novel splice events. Most common approach.
- align reads to transcriptome index (required transcripts to be known and annotated in the reference) - i.e. pseudoalignment. Use if you have short reads < 50bp. Using Kallisto or Salmon - much quicker but wont identify new transcripts.
- de novo assembly: assemble transcripts from overlapping tags. Useful if no reference genome exists for species studied. Also if looking for complex polymorphisms that would be missed by aligning to reference.

Aligning to Genome vs de-novo assembly:

~ 2005 high throughput short-read sequencers changed the face of sequencing which became much cheaper and accessible. Previously sequencing was laborious and expensive and the focus was on producing accurate long reads (1000bp). Sequencing reads longer improves alignment. Sequencers now produce millions of short reads (50-300bp). Aligners have thus changed to adapt to short reads - rapidly select the best mapping at the expense of not investigating all potential alternative alignments.
- Short read aligners are incredible! They match > 10,000 sequences per second against 3 billion bases of the human genome.
- There is large variation in results between different aligners. A tool that prioritises finding exact matches will do so at the expense of missing locations and overlaps and vice versa.
- Limitations:
- finds alignments that are reasonably similar but not exact (algorithm will not search beyond a defined matching threshold)
- cannot handle very long reads or very short reads (<30bp) (become inefficient)
Aligners aim to:
- speed up process
- reduce amount of memory used
- maximise quality of the alignment
Note the RNA aligners in red below:

The main RNA-seq aligner milestones are:
- TopHat > TopHat2. Slowish 1000mins. Low memory 4GB. Everyone used it from 2009
- STAR. Faster 24mins. Used more memory 28GB (need a server)
- HISAT. Faster 20mins & reduce memory 4GB
| Aligner | Speed | Memory | Year |
|---|---|---|---|
| TopHat > TopHat2 | 1000mins | 4GB | 2009 |
| STAR | 24mins | 28GB | 2012 |
| HISAT > HISAT2 | 20mins | 4GB | 2016 |
If you are aligning to a genome then you need a splice aware aligner:
- reads span large introns
- reads represent mature mRNA have introns removed
- but aligning back to the reference genome needs to recognise intron sites
- unless reads a short <50bp then you need a splice aware aligner

Options:
- STAR: Really fast, produces counts for you too. Straight forward RNA seq for differential gene expression analysis. Efficient. Sensitivie. Identified large number of novel splice sites.
- HISAT2: similar algorithms as Bowtie2/Tophat2 - but performs at much faster speeds
- Subjunc: designed specifically for gene expression analysis.
- BWA: the MEM algorithm of bwa supports spliced alignments.
- BBMap: Is capable of detecting very long splicing.
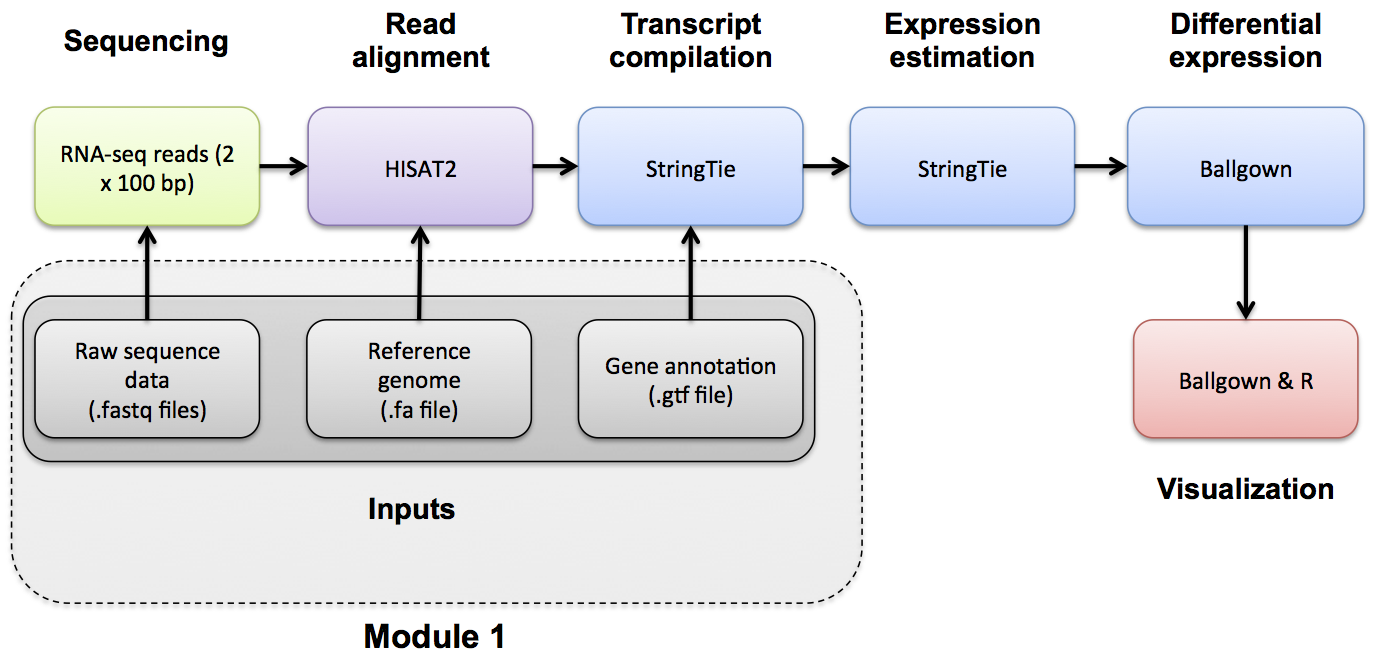
- Tuxedo suite refers to the pioneering automating pipelines for RNA-seq = alignment with tophat & bowtie, and Quantification & Differential Expression with cufflinks - all now outdated.
- The new Tuxedo suite refers to
- alignment: hisat2
- quantification: stringtie
- differential expression: ballgown (R package)
The aligners first align to a global index (slow) & then to a local index (faster):
- global indexing: tries to anchor the first 28 bp of the read to the reference genome > find the global location in the genome of where the read belongs. Then extends the anchor base by base. If the read crosses a splice junction it will encounter a mismatch > switch to local index.
- local indexing: align remaining bases to the downstream or upstream exon.

When choosing an aligner, we need to decide what features the aligner should provide:
- Alignment algorithm: global, local, or semi-global?
- Is there a need to report non-linear arrangements?
- How will the aligner handle INDELs (insertions/deletions)?
- Can the aligner skip (or splice) over large regions?
- Can the aligner filter alignments to suit our needs?
- Will the aligner find chimeric alignments?
Reference genome sequence repositories: GenCODE - Generally for human data use ENSEMBL : has the most detailed annotations of genomes. UCSC table browser - Stores large scale result tiles produced by consortia e.g. ENCODE. Contains multiple sequence alignment datasets across entire genomes.https://genome.ucsc.edu/cgi-bin/hgTables ENCODE iGenomes NCBI Mouse Genome Project Berkeley Drosphilia Project RefSeq RNA-Central - non coding RNA sequencing database
- UCSC and Ensembl use different naming conventions (which impacts on analyses) - try to stick to one.
- must use Ensembl FASTA file with Ensemble GTF file. You cannot mix Ensembl with UCSC without modifying first and this isn't advised as they have different scaffolds.
Reference sequences are FASTA files. Reference sequences are long strings of ATCGN letters.
Annotation files store start sites, exon, introns. They contain spacial coordinate information describing genomic loci (2D position) according to their pattern (chromosome, start, end) as intergers. These integers represent the left & right positions of an interval on the genome.
Both formats contain information:
- Span: start & end coordinates
- Hierarchy: feature belongs to another feature e.g. exon to a transcript
- Value: at a sapsecific coordinate
- Annotation: feature X is also characterised by Y & Z
2 types of genome formats exist:
BED Format BED is 0 based, non-inclusive on the right. This means that the interval [1, 5) contains 1,2,3,4 and coordinate 1 is the second coordinate of the genome (0 is the first). A single line record stores all the information on the block structure of a record.
3 compulsory fields: chromosome & start & end. 9 optional fields: name, score, strand, thickStart, thickEnd, itemRgb, blockCount, blockSizes, blockStarts field number can vary from 3 - 12. Must be consistent within a file.
RSeQC requires BED format.
GFF = General Feature Format GFF is 1 based, inclusive on both ends. This means that [1,5] contains 1,2,3,4,5 and that coordinate 1 is the first coordinate of the genome. The relationship is built from data distributed over multiple lines.
9 fields separated by TAB.
- Reference sequence: coordinate system of annotation eg Chr1
- Source: annotation software
- Method: annotation type (eg gene) [GFF3 = Type: term from lite Sequence Ontology SOFA or accession number]
- Start Position: 1-based integer, <= stop position
- Stop Position: 0-length features (insertion sites)
- Score: sequence identity
- Strand: + = forward. - = reverse. “.” = no stranded
- Phase: codon phase for annotations linked to proteins. 0; 1; or 2 = indicates frame (bases to be removed from beginning of feature to reach first base of next codon)
- Group: class and ID of an annotation which is the parent of the current one. [GFF3 = Attributes: TAG=VALUE pairs - ID, name, alias, parent, target, gap]
- Source
- Type
Always use GFF based formats. Can convert between the two formats using:
ml BEDOPS
GTF=/camp/home/ziffo/home/genomes/annotation/GRCh38.p12/gencode.v28.primary_assembly.annotation.gtf
BED=/camp/home/ziffo/home/genomes/annotation/GRCh38.p12/gencode.v28.primary_assembly.annotation.bed
awk '{ if ($0 ~ "transcript_id") print $0; else print $0" transcript_id \"\";"; }' $GTF | gtf2bed - > $BEDml BEDOPS
GTF=/camp/home/ziffo/home/genomes/annotation/Homo_sapiens.GRCh38.99.gtf
BED=/camp/home/ziffo/home/genomes/annotation/Homo_sapiens.GRCh38.99.bed
awk '{ if ($0 ~ "transcript_id") print $0; else print $0" transcript_id \"\";"; }' $GTF | gtf2bed - > $BED
Keep reference genomes in a more general location rather than the local folder, since you will reuse them frequently: /camp/home/ziffo/home/genomes/ and place into repository subdirectories:
gencode/
ensembl/
ucsc/
https://www.gencodegenes.org/human/
Downloasequencesand then make a new directorymkdir`
GENCODE process: https://www.gencodegenes.org/releases/current.html find the latest human reference genome: currently this is Release 3328 (GRCh38.p13).2) Copy the Link address to download the FASTA file to the GENOME SEQUENCE PRI (primary) - bottom of 2nd table. I Then in command line download to the appropriate directory:
cd ~/working/oliver/genomes/gencode
#wget [paste link address]
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_33/GRCh38.primary_assembly.genome.fa.gz
# gunzip filename
gunzip GRCh38.primary_assembly.genome.fa.gzDownload the GTF file to the Comprehensive gene annotation PRI (primary) regions:
cd ~/working/oliver/genomes/gencode
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_33/gencode.v33.primary_assembly.annotation.gtf.gz
gunzip gencode.v33.primary_assembly.annotation.gtf.gz
chmod 777 gencode.v33.primary_assembly.annotation.gtf.gz gencode.v33.primary_assembly.annotation.gtf.gzwget [paste link address] then gunzip filename
Do the same for the GTF file to the Comprehensive gene annotation PRI (primary) regions.
ENSEMBL process: http://www.ensembl.org/info/data/ftp/index.html Search for species of interest Click on Gene sets GTF link link & DNA (FASTA) link. NB cDNA is transcriptome (not genome).
FASTA: Right click on dna.primary_assembly.fa link GTF: Right click on Saccharomyces)cerevisiae.R64-1-1.92.gtf.gz → copy link address e.g. ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
GTF
FASTA: Right click on Human → cDNA topy link address for largest gtf chr_patch_hapl_scaff e.g. ftp://ftp.ensembl.org/pub/release-99/gtf/homo_sapiens/Homo_sapiens.GRCh38.99.chr_patch_hapl_scaff.gtf.gz
In command line (in appropriate Folder) wget [paste link address]
Unzip file gunzip file_name
cd ~/working/oliver/genomes/ensembl
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/gtf/homo_sapiens/Homo_sapiens.GRCh38.99.chr_patch_hapl_scaff.gtf.gz
gunzip Homo_sapiens.GRCh38.99.chr_patch_hapl_scaff.gtf.gz
chmod 777 Homo_sapiens.GRCh38.99.gtf Homo_sapiens.GRCh38.dna.primary_assembly.faIn command line (in appropriate Folder) wget [paste link address]
Unzip file gunzip file_name
UCSC process
:
ml Kent_tools
Download a GTF file of yeast transcripts from the UCSC Genome Table Browser https://genome.ucsc.edu/cgi-bin/hgTables
Move the downloaded GTF file to the appropriate folder

remove first column & first line: cut -f 2- file_name.txt | sed ‘1d”
genePredToGtf file file_name file_name.gtf
Compress with gzip command or faToTwoBit filename.fa filename.2bit. Can then reconvert 2bit format —> FASTA format: twobittofa file_name.2bit file_name.fa
- The aim of alignment is to produce a SAM (Sequence Alignment Map) file which contains all information on the sequencing & its alignment. After creating the SAM file there is no need to look at the FASTQ file again since the SAM contains all the FASTQ information.
The index often needs updating to match the current version of STAR.
- All short read aligners first build an index from the reference genome. Then the FASTQ sequencing files are aligned against this index.
- Index building prepares the reference genome to allow the tools to search it efficiently. It creates multiple files that should be stored near to the FASTA reference sequence.
- Can take days for large genomes to build an index. The relevant indexes have already been created and stored on the CAMP cluster in
/home/camp/ziffo/working/luscombelab-UCL
Input Files = Reference genome sequence (FASTA) & Annotation file (GTF)
Create directory to store index in: mkdir GRCh38.12_STAR_index
Module load STAR ml STAR
STAR commands have the format: STAR --option1-name option1-value(s)--option2-name option2-value(s) ...
To generate the index in STAR, specify the location of:
- index directory to store the output
- FASTA reference genome file
- GTF annotation file
- Overhand: read length minus 1. Read length distribution are shown in the MultiQC report. This is length of the genomic sequence around the annotated junction to be used for the splice junctions database
ml STAR # version 2.6.1 noted in Log.out file
cd /camp/lab/luscomben/home/users/ziffo/genomes/ensembl/GRCh38.99.STAR_index
OUT=/camp/lab/luscomben/home/users/ziffo/genomes/ensembl/GRCh38.99.STAR_index
FASTA=/camp/lab/luscomben/home/users/ziffo/genomes/ensembl/Homo_sapiens.GRCh38.dna.primary_assembly.fa
GTF=/camp/lab/luscomben/home/users/ziffo/genomes/ensembl/Homo_sapiens.GRCh38.99.gtf
#Send cmd to generate index as batch job to cluster:
sbatch -N 1 -c 10 --mem=80G -t 48:00:00 --wrap="STAR --runMode genomeGenerate --genomeDir $OUT --genomeFastaFiles $FASTA --sjdbGTFfile $GTF --sjdbOverhang 99 --runThreadN 20 --limitGenomeGenerateRAM 170263683456" --mail-type=ALL,ARRAY_TASKS --mail-user=oliver.ziff@crick.ac.uk think of --sjdbOverhang as the maximum possible overhang for your reads. Ideally 1- read length"
The above code is resuable and applicable to all situations by editing the $changable elements.
The "hard-coding" looks like this (very long and difficult to edit):
```bash
sbatch -N 1 -c 8 --mem 40G --wrap="STAR --runMode genomeGenerate --genomeDir --genomeFastaFiles /camp/home/ziffo/home/genomes/sequences/human/GRCh38.primary_assembly.genome.fa --sjdbGTFfile /camp/home/ziffo/home/genomes/annotation/GRCh38.p12/gencode.v28.primary_assembly.annotation.gtf --sjdbOverhang 959 --runThreadN 8"
Used with rMATS version 2.4.1
STAR/2.7.7a-GCC-10.2.0
ml STAR/2.7.7a-GCC-10.2.0 # version 2.7.7 noted in Log.out file
mkdir /camp/lab/luscomben/home/users/ziffo/genomes/ensembl/GRCh38.99.STAR.2.7.7_index
cd /camp/lab/luscomben/home/users/ziffo/genomes/ensembl/GRCh38.99.STAR.2.7.7_index
OUT=/camp/lab/luscomben/home/users/ziffo/genomes/ensembl/GRCh38.99.STAR.2.7.7_index
FASTA=/camp/lab/luscomben/home/users/ziffo/genomes/ensembl/Homo_sapiens.GRCh38.dna.primary_assembly.fa
GTF=/camp/lab/luscomben/home/users/ziffo/genomes/ensembl/Homo_sapiens.GRCh38.99.gtf
#Send cmd to generate index as batch job to cluster:
sbatch -N 1 -c 10 --mem=80G -t 4:00:00 --wrap="STAR --runMode genomeGenerate --genomeDir $OUT --genomeFastaFiles $FASTA --sjdbGTFfile $GTF --sjdbOverhang 99 --runThreadN 20 --limitGenomeGenerateRAM 170263683456" --mail-type=ALL,ARRAY_TASKS --mail-user=oliver.ziff@crick.ac.uk Gencode index GRCh38.p13 v33 with STAR version 7.1
ml STAR SAMtools
cd /camp/home/ziffo/home/genomes/gencode/GRCh38.p13_STAR_index
OUT=/camp/home/ziffo/home/genomes/gencode/GRCh38.p13_STAR_index
FASTA=/camp/home/ziffo/home/genomes/gencode/GRCh38.p13.primary_assembly.genome.fa
GTF=/camp/home/ziffo/home/genomes/gencode/gencode.v33.primary_assembly.annotation.gtf
#Send cmd to generate index as batch job to cluster:
sbatch -N 1 -c 8 --mem=0 -t 48:00:00 --wrap="STAR --runMode genomeGenerate --genomeDir $OUT --genomeFastaFiles $FASTA --sjdbGTFfile $GTF --sjdbOverhang 99 --runThreadN 8 --limitGenomeGenerateRAM 170263683456" --mail-type=ALL,ARRAY_TASKS --mail-user=oliver.ziff@crick.ac.uk
```Check it is running using `myq`
If it isn’t seen there then `ls` the folder → look for output file named “slurm…”
Open slurm file: `more slurm…` which will explain outcome of file eg FATAL INPUT PARAMETER ERROR
### 2. Align each FASTQ file to the Index
ml STAR
ml Python/3.5.2-foss-2016b
ml SAMtools
* Sample distributed over X flow cell lanes → X fastq files per sample
* STAR merges the X files if multiple file names are indicated (other align tools don't)
* Separate the file names with a comma (no spaces)
* Create directory to store STAR output `mkdir alignment`
By allocating all file names to the `$INPUT` term it means all the FASTQ files are read together (see example further down) but it is best to do each separately - can use [snakemake](http://slides.com/johanneskoester/snakemake-tutorial#/) - this makes it easier to see if there is an alignment error in each individual sequencing file:
* List fast.qz files separated by commas and remove white spaces:
`INPUT= ls -m /camp/home/ziffo/home/projects/airals/fastq_files/SRR5*.fastq| sed 's/ //g' | echo $INPUT | sed 's/ //g'`
`ls -m` list, fill width with a comma separated list of entries all the fastq files.
`sed` remove a space from each file name
`echo` displays a line of text - in this case it lists all the file names - in this case the pipe to echo is to remove new spaces that are created between multiple lines. no space after FILES - with space after it thinks FILES is command.
`sed` = stream editor - modify each line of a file by replacing specified parts of the line. Makes basic text changes to a file - `s/input/output/g`
```bash
#set the index
IDX=/camp/home/ziffo/home/genomes/index/GRCh38.p12_STAR_index
#set the fastq sequencing file to read in
READ1=/camp/home/ziffo/home/projects/airals/reads/D0_samples/trimmed_depleted/*.sam
#set the paired fastq sequencing file to read in (for paired end data only)
READ2=
#set name under which to store the BAM file output
BAM=/camp/home/ziffo/home/projects/airals/alignment_STAR/D7_samples/trimmed_filtered_depleted/SRR5483788_
#SEND ALIGNMENT AS SBATCH
sbatch -N 1 -c 8 --mem 40G --wrap="STAR --runThreadN 1 --genomeDir $IDX --readFilesIn $READ1 --outFileNamePrefix $BAM --outFilterMultimapNmax 1 --outSAMtype BAM SortedByCoordinate --outReadsUnmapped Fastx --twopassMode Basic"The above script needs editing for each sequencing file (redundant). To create a script that runs all sequencing files (non-redundant) you can use a for loop or snakemake.
For Loop
# create all time point output folders
mkdir D0_samples D7_samples D14_samples D21_samples D35_samples D112_samples
#set the index
IDX=/camp/home/ziffo/home/genomes/index/GRCh38.p12_STAR_index
# set timepoint folders
TIMEPOINT=/camp/home/ziffo/home/projects/airals/reads/D*_samples
## run multiple alignments using in for loop
for SAMPLE in $TIMEPOINT;
do
DAY=`echo $SAMPLE | grep -E -o 'D[0-9]+_samples'`
#set the sequencing file to read in (use trimmed_depleted output)
READ1=$SAMPLE/trimmed_depleted/*.fastq
for REPLICATE in $READ1
do
#set BAM output file aligned to human genome
BAM=/camp/home/ziffo/home/projects/airals/alignment/$DAY
SRRID=`echo $REPLICATE | grep -E -o 'SRR[0-9]+'`
sbatch -N 1 -c 8 --mem 40G --wrap="STAR --runThreadN 1 --genomeDir $IDX --readFilesIn $REPLICATE --outFileNamePrefix ${BAM}/${SRRID}_ --outFilterMultimapNmax 1 --outSAMtype BAM SortedByCoordinate --outReadsUnmapped Fastx --twopassMode Basic"
# Index each BAM file as they are produced
samtools index ${BAM}/${SRRID}_Aligned.sortedByCoord.out.bam
done
doneBiostars approach to alignment:
# Create output folder
mkdir -p bam
# set the index
IDX=/camp/home/ziffo/home/genomes/index/GRCh38.p12_STAR_index
for SAMPLE in VCP CTRL;
do
for REPLICATE in 1 2 3;
do
# Build the name of the files.
READ1=reads/${SAMPLE}_${REPLICATE}_R1.fq
BAM=bam/${SAMPLE}_${REPLICATE}.bam
echo "Running STAR on $SAMPLE"
# Run the aligner.
STAR --runThreadN 1 --genomeDir $IDX --readFilesIn $READ1 --outFileNamePrefix $BAM --outFilterMultimapNmax 1 --outSAMtype BAM SortedByCoordinate --outReadsUnmapped Fastx --twopassMode Basic
# Index each BAM file as they are produced
samtools index $BAM
done
done
#Place the above into a script with atom & save in appropriate folder, name it `align.sh` and run it with
bash align.shSnakefile
- write the following rules in Atom and save the file in the appropriate directory
rule star_map:
input:
index="/camp/home/ziffo/home/genomes/index/GRCh38.p12_STAR_index"
sample="/camp/home/ziffo/home/projects/airals/fastq_files/{sample}.fastq",
output:
"/camp/home/ziffo/home/projects/airals/alignment_STAR/{sample}.sam"
shell:
"STAR --genomeDir {input.index} --readFilesIn {input.sample} --outFileNamePrefix {output}_ --outFilterMultimapNmax 1 --outReadsUnmapped Fastx --outSAMtype BAM SortedByCoordinate --twopassMode Basic --runThreadN 1"
#dry run workflow:
`snakemake -np /camp/home/ziffo/home/projects/airals/alignment_STAR/{sample}.sam`
#execute workflow:
`snakemake /camp/home/ziffo/home/projects/airals/alignment_STAR/{sample}.sam`The STAR manual has all the explanations on how to write the STAR command and fine tune parameters including:
- multi-mapped reads
- optimise for small genomes
- define min & max intron sizes
- handle genomes with >5000 scaffolds
- detect chimeric & circular transcripts
if files are compressed add
--readFilesCommand gunzip -cSTAR ENCODE options:outFilterMultimapNmax 1max number of multiple alignments allowed for a read - if exceeded then read is considered unmapped i.e. when 1 is used this only identifies unique reads and removes multimapped reads. This is generally accepted.
#STAR will perform the alignment, then extract novel junctions which will be inserted into the genome index which will then be used to re-align all reads
#runThreadN can be increased if sufficient computational power is available
STAR Output Files
- Aligned.sortedByCoord.out.bam - the loci of each read & sequence
- Log.final.out - summary of alignment statistics
- Log.out - commands, parameters, files used
- Log.progress.out - elapsed time
- SJ.out.tab - loci where splice junctions were detected & read number overlapping them
- Unmapped.out.mate1 - fastq file with unmapped reads
-
check the slurm file:
more slurm-132XXX.out -
check the summary of the output:
cat FILENAME_Log.final.out
-
check that the number of reads in the fastq files matches the STAR log output:
more SRR5483788_Log.final.outecho $( cat /camp/home/ziffo/home/projects/airals/fastq_files/D7_samples/SRR5483789_1.fastq | wc -l)/4 | bc
ml SAMtools
- STAR auto creates BAM files.
-bwill produce a BAM file.-swill produce a SAM file. - Now need to index each BAM file. The indexed BAM file format = BAM.BAI file. Inxed allows quick access to the BAM files without having to load them to memory.
for individual files: samtools index filename_Aligned.sortedByCoord.out.bam
Using the STAR alignment For Loop above you can automate this with the above phase as each BAM file is produced:
for file in /camp/home/ziffo/home/projects/airals/alignment/D7_samples/trimmed_filtered_depleted/SRR5*_Aligned.sortedByCoord.out.bam
do
sbatch -N 1 -c 8 --mem 40 --wrap="samtools index $file";
done# set timepoint folders
TIMEPOINT=/camp/home/ziffo/home/projects/airals/reads/D*_samples
for SAMPLE in $TIMEPOINT;
do
READ=$SAMPLE/trimmed_depleted/*.fastq
for REPLICATE in $READ
do
SRRID=`echo $REPLICATE | grep -E -o 'SRR[0-9]+'`
BAM=$SAMPLE/${SRRID}Aligned.sortedByCoord.out.bam
samtools index $BAM
done
doneSAM/BAM files store read alignments.
SAM files are generic nucleotide alignment format describing alignment of sequenced reads to a reference. SAM format are TAB-delimited line-orientated (each row represents a single read alignment) text consisting of 2 types of tags:
- Header: meta-data
- Body: longer with information on alignment.
- The SAM format specification is the official specification of the SAM format.
- The SAM tag specification is the official specification of the SAM tags.
- impossible to transform one aligners SAM file into another aligners SAM file.
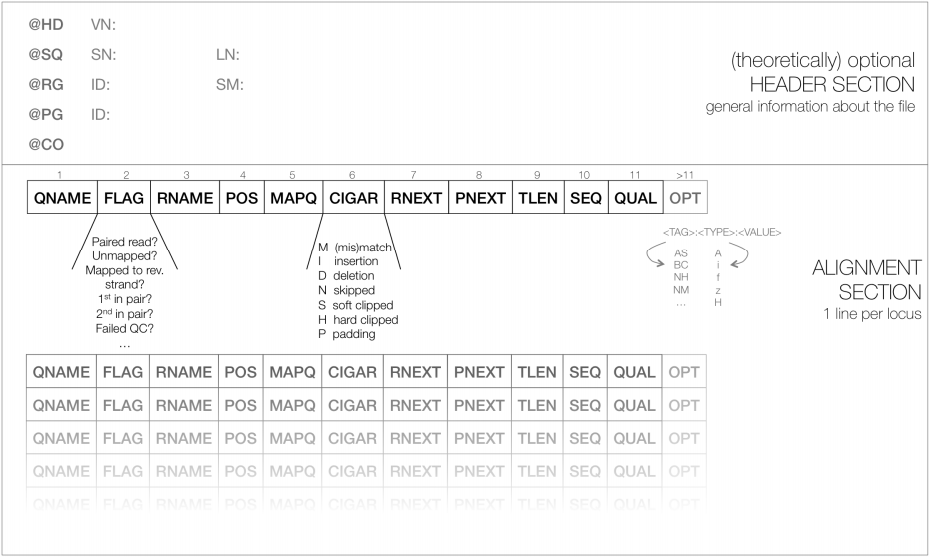
Describes the data source, reference sequence, method of alignment.
Each section begins with @then value pairs 2 letter abbreviations:
@HD = header line: VN version format, SO sorting order of alignments
@SQ = sequence directory listing SN sequence name aligned against (from FASTA), LN sequence length, SP species,
@RG Read Group = ID read group identifier, CN name of sequencing centre, SM sample name
@PG program ran = PN programme name, VN program version
- 1 line per chromosome
- To retrieve only the SAM header
samtools view -H - To retrieve both the header & alignment sections
samtools view -h - The default
samtools viewwill not show the header section - Print read group tags
samtools view -H filename.bam | cut -f 12-16 | head -1. You may need to add a custom read group tag in individual bam files prior to merging withsamtools addreplacergusingTAG:FORMAT:VALUE. This allows pooling results of multiple experiments into a single BAM dataset to simplify downstream logistics into 1 dataset.samtools view -H filename.bam
- Each line corresponds to one sequenced read.
- The SAM tags are defined here
- Each SAM file has 11 mandatory columns. Despite this aligners vary in how much information they put into these columns:
<QNAME> <FLAG> <RNAME> <POS> <MAPQ> <CIGAR> <MRNM> <MPOS> <ISIZE> <SEQ> <QUAL>

- Following the 11 mandatory fields, the optional fields are presented as key-value pairs in the format of
<TAG>:<TYPE>:<VALUE>, whereTYPEis one of:A- Characteri- Integerf- Float numberZ- StringH- Hex string - Reads within the same SAM file may have different numbers of optional fields, depending on the aligner program that generated the SAM file.
- compresses all mapping information on a read alignment into one single decimal number
- decimal is the sum of all the answers to Yes/No questions:
Binary Integer Name Meaning
000000000001 1 PAIRED Read paired
000000000010 2 PROPER_PAIR Read mapped in proper pair
000000000100 4 UNMAP Read unmapped
000000001000 8 MUNMAP Mate unmapped
000000010000 16 REVERSE Read reverse strand
000000100000 32 MREVERSE Mate reverse strand
000001000000 64 READ1 First in pair, file 1
000010000000 128 READ2 Second in pair, file 2
000100000000 256 SECONDARY Not a primary alignment
001000000000 512 QCFAIL Read fails platform/vendor quality checks
010000000000 1024 DUP Read is PCR or optical duplicate
100000000000 2048 SUPPLEMENTARY Supplementary alignment
- To decode the FLAG integer into plain english click here.
samtools flagstat assesses the FLAG field and prints a summary report:
samtools flagstat Aligned.sortedByCoord.bam
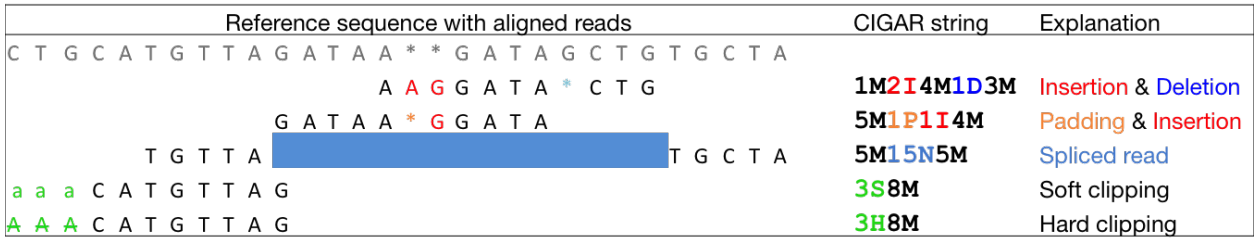
CIGAR string letters M, I, D etc to indicate how the read aligned to the reference sequence.

The sum of lengths of the M, I, S, =, X operations must equal the length of the read. Here are some examples:
For each base alignment the options are:
- - a space
- | match
- . a mismatch
Scoring is based on the value you associate with a match, mismatch or a space. Alignment algorithms find the arrangement that produce the maximal alignment score. Example scoring matrix: - 5 points for a match. - -4 points for a mismatch. - -10 points for opening a gap. - -0.5 points for extending an open gap.
- Modifying the scoring algorithm can dramatically change the way that the sequence is aligned to the reference. See this example where reducing the gap penalty from -10 to -9 actually corrects the alignment. This setting means that 2 matches (5 + 5 = 10) overcomes a penality gap open (9). Thus to achieve a higher overall score the aligner will prefer opening a gap anytime it can find two matches later on (net score +1).
- EDNAFULL is the default scoring choice for all aligners.
- BAM file = Binary Alignment Map - human readable TAB-delimited compressed. BAM files are binary, compressed and sorted representation of SAM information with alignment coordinates allowing fast query on info by location. Used to exchange data as it quicker than SAM.
- Bigger than gzipped SAM files as they are optimised for rapid access (not just size reduction). SAM files are human readable, BAM are compressed. BAM are much smaller.
- for SAM files you can run other commands on them eg head FILENAME.sam whereas BAM files need to be run through samtools i.e.
samtools view FILENAME.bam | cut -f 2 | head
#set BAM input files
BAM=/camp/home/ziffo/home/projects/airals/alignment/D7_samples/trimmed_filtered_depleted/SRR54837*_Aligned.sortedByCoord.out.bam
#set output file directory
OUT=/camp/home/ziffo/home/projects/airals/alignment/D7_samples/trimmed_filtered_depleted/
#run samtools on each BAM file separately
for SAMPLE in $BAM
do
SRRID=`echo $SAMPLE | grep -E -o 'SRR[0-9]+'`
sbatch -N 1 -c 8 --mem=40GB --wrap="samtools view -h $SAMPLE > ${SRRID}.sam"
done
# set timepoint folders
TIMEPOINT=/camp/home/ziffo/home/projects/airals/alignment/D*_samples
for SAMPLE in $TIMEPOINT;
do
BAM=$SAMPLE/*Aligned.sortedByCoord.out.bam
echo "Running timepoint $SAMPLE"
for REPLICATE in $BAM
do
SRRID=`echo $REPLICATE | grep -E -o 'SRR[0-9]+'`
sbatch -N 1 -c 8 --mem=40GB --wrap="samtools view -h $REPLICATE > $SAMPLE/${SRRID}.sam"
echo "Running sample $SRRID"
done
doneor alternatively:
samtools view -h FILENAME_Aligned.sortedByCoord.out.bam > FILENAME_Aligned.sortedByCoord.out.sam
Convert a BAM file into a SAM file (including the header): samtools view -h FILENAME.bam > FILENAME.sam
Compress a SAM file into BAM format (-Sb = -S -b)" samtools view -Sb FILENAME.sam > FILENAME.bam
To peak into a SAM or BAM file: samtools view FILENAME.bam | head
BAM files can be massive. Often we are only interested in a focused subset of the reference genome. These region of interest subsets are speficied in BED files. To make files smaller you can use only the BED file which are accepted by BAM manipulation tools eg SAMTools, piccard
Similar to BAM (binary compressed) but smaller as some compression is in the reference genome. Sometimes you need the reference genome information so these arnt always appropriate. Supported by samtools Concerted effort to move from BAM to CRAM.
There are 4 major toolsets for processing SAM/BAM files:
- Picard - Java tools for manipulating high throughput sequencing data
- BAMtools - read, write & manipulate BAM genome alingment files .
ml BamTools - SAMTools - interact with high throughput sequencing data, manipulate alignments in SAM/BAM format, sort, merge, index, align in per-position format. SAMTools help page =
samtools --helpUsage:samtools <command> [options]
5 key SAMTool commands:
- Indexing
- Editing
- File operations (aligning, converting, merging)
- Statistics
- Viewing
- BAM files are usually sorted by position coordinates. This allows rapid access to the huge BAM file.
- Some tools require BAM files to be sorted by read name usually when we need to identify both reads of a pair.
Unlike other aligners, STAR already creates separate bam files of aligned and unmapped.
Use FLAGS to filter using samtools: -f flag includes matches; -F flag includes mismatches. Flag 4 indicates unmapped.
Count alignments that were aligned: samtools view -c -F 4 filename.bam
Get overview of alignments:
report of flags samtools flagstat filename.bam
report on how many reads align to each chromosome samtools idxstats filename.bam
report on flags bamtools stats -in filename.bam
Calculate the depth of coverage: samtools depth filename.bam | head or bamtools coverage -in filename.bam | head
Count reads that have multiple alignments samtools view -c -F 4 -f 256 filename.bam
Count supplementary (chimeric) alignements samtools view -c -F 4 -f 2048 filename.bam
Count primary (non supplementary & non secondary) alignments samtools view -c -F 4 -F 2304 filename.bam
Filter on mapping quality using -q flag:
Count number of reads with mapping quality >20
samtools view -c -q 20 filename.bam
Create bam file with mapping quality (Phred Scale) >= 20
samtools view -h -b -q 20 FILENAME.bam > high_mapq_reads.bam
Create BAM with uniquely aligned reads (STAR gives uniquely aligned reads a mapping quality of 255 so pull reads with mapping quality = 255 samtools view -h -q 255 FILENAME.bam > uniquely_aligned_reads.bam .
Create BAM file with only reads aligned to reverse strand:
- sort the BAM file (A-Z; 0-9) and count the adjacent lines which are identical using
samtools view FILENAME.bam | cut -f 2 | sort | uniq -c - Then, create file with the specific feature e.g. reverse reads (FLAG = 16 in column 2):
samtools view -h -f 16 FILENAME.bam > reverse_reads.bam
Create SAM file with reads of insert sizes > 1000bp use the CIGAR string (column 6 in SAM file):
- first convert BAM to SAM file
samtools view -h FILENAME.bam > FILENAME.sam - use
grepto exclude (using-v) lines with >3 digits (using[0-9][0-9][0-9][0-9]) followed byN(N means mismatch i.e. skipped bases, whereas M = match)egrep -v "[0-9][0-9][0-9][0-9]N" FILENAME.sam > smallinsert_reads.sam - Alternatively use
awkto focus on column 6 ($6in CIGAR string) and exclude lines with 3 digits (using![0-9][0-9][0-9][0-9]) then printing everything{print $0}and creating new file:awk '!($6 ~ /[0-9][0-9][0-9][0-9]N/) {print $0}' FILENAME.sam > smallinsert_reads.sam
Create SAM file with intron spanning reads:
- use
grepto select lines with a number of digits (using[0-9]+) thenM(i.e. matches) then any number of digits again, thenN(i.e. mismatches) then any number of digits and then M again at the end:egrep "(^@|[0-9]+M[0-9]+N[0-9]+M)" FILENAME.sam > intron-spanning_reads.sam - Alternatively use awk to focus on column 6 (CIGAR string) and select the header
$1 ~ /^@/and the 6th column with any number of digits followed by M followed by digits then N then digits the M:awk '$1 ~ /^@/ || $6 ~ /[0-9]+M[0-9]+N[0-9]+M/ {print $0}' FILENAME.sam > intron-spanning_reads.sam
As you aligned each fastq file separately you have a BAM file for each fastq. At some point you will need to merge the BAM files for downstream processing.
You can merge BAM files from each condition (VCP or CTRL) but not combine different conditions (wildtype & mutant). Dont forget there maybe batch effects between samples in the sample condition which are difficult to analyse after merging BAM files.
#run samtools merge command listing all BAM files to read in
samtools merge all_bam_files.bam filename1.bam filename2.bam filename3.bam`
#Check the new merged bam file
samtools view -H all_bam_files.bam
#index the new merged bam file (creates the BAI file which is required for downstream analysis)
samtools index all_bam_files.bamml MultiQC
Generate a comprehensive & interactive report of STAR alignment Log.final.out files using MultiQC
Go to the alignment folder with the STAR Log.final.out files in and run: multiqc .
Interpret the HTML report.
Compare the alignment MultiQC HTML reports (the raw unprocessed aligned read report & the trimmed, filtered & depleted aligned read report)