No template found for residue #2103
Comments
|
Here are the PDB file contents: And I've put the other one here as to not clutter this page |
|
It looks like your nucleotide is terminated differently from what's defined in the force field. Amber14 requires nucleotides to be terminated in one specific way, and doesn't include parameters for any other form. Here is its definition of an isolated DT: <Residue name="DTN">
<Atom charge="0.4422" name="HO5'" type="DNA-HO"/>
<Atom charge="-0.6318" name="O5'" type="DNA-OH"/>
<Atom charge="-0.0069" name="C5'" type="DNA-CJ"/>
<Atom charge="0.0754" name="H5'" type="DNA-H1"/>
<Atom charge="0.0754" name="H5''" type="DNA-H1"/>
<Atom charge="0.1629" name="C4'" type="DNA-CT"/>
<Atom charge="0.1176" name="H4'" type="DNA-H1"/>
<Atom charge="-0.3691" name="O4'" type="DNA-OS"/>
<Atom charge="0.068" name="C1'" type="DNA-CT"/>
<Atom charge="0.1804" name="H1'" type="DNA-H2"/>
<Atom charge="-0.0239" name="N1" type="DNA-N*"/>
<Atom charge="-0.2209" name="C6" type="DNA-CM"/>
<Atom charge="0.2607" name="H6" type="DNA-H4"/>
<Atom charge="0.0025" name="C5" type="DNA-CM"/>
<Atom charge="-0.2269" name="C7" type="DNA-CT"/>
<Atom charge="0.077" name="H71" type="DNA-HC"/>
<Atom charge="0.077" name="H72" type="DNA-HC"/>
<Atom charge="0.077" name="H73" type="DNA-HC"/>
<Atom charge="0.5194" name="C4" type="DNA-C"/>
<Atom charge="-0.5563" name="O4" type="DNA-O"/>
<Atom charge="-0.434" name="N3" type="DNA-NA"/>
<Atom charge="0.342" name="H3" type="DNA-H"/>
<Atom charge="0.5677" name="C2" type="DNA-C"/>
<Atom charge="-0.5881" name="O2" type="DNA-O"/>
<Atom charge="0.0713" name="C3'" type="DNA-C7"/>
<Atom charge="0.0985" name="H3'" type="DNA-H1"/>
<Atom charge="-0.0854" name="C2'" type="DNA-CT"/>
<Atom charge="0.0718" name="H2'" type="DNA-HC"/>
<Atom charge="0.0718" name="H2''" type="DNA-HC"/>
<Atom charge="-0.6549" name="O3'" type="DNA-OH"/>
<Atom charge="0.4396" name="HO3'" type="DNA-HO"/>
<Bond atomName1="HO5'" atomName2="O5'"/>
<Bond atomName1="O5'" atomName2="C5'"/>
<Bond atomName1="C5'" atomName2="H5'"/>
<Bond atomName1="C5'" atomName2="H5''"/>
<Bond atomName1="C5'" atomName2="C4'"/>
<Bond atomName1="C4'" atomName2="H4'"/>
<Bond atomName1="C4'" atomName2="O4'"/>
<Bond atomName1="C4'" atomName2="C3'"/>
<Bond atomName1="O4'" atomName2="C1'"/>
<Bond atomName1="C1'" atomName2="H1'"/>
<Bond atomName1="C1'" atomName2="N1"/>
<Bond atomName1="C1'" atomName2="C2'"/>
<Bond atomName1="N1" atomName2="C6"/>
<Bond atomName1="N1" atomName2="C2"/>
<Bond atomName1="C6" atomName2="H6"/>
<Bond atomName1="C6" atomName2="C5"/>
<Bond atomName1="C5" atomName2="C7"/>
<Bond atomName1="C5" atomName2="C4"/>
<Bond atomName1="C7" atomName2="H71"/>
<Bond atomName1="C7" atomName2="H72"/>
<Bond atomName1="C7" atomName2="H73"/>
<Bond atomName1="C4" atomName2="O4"/>
<Bond atomName1="C4" atomName2="N3"/>
<Bond atomName1="N3" atomName2="H3"/>
<Bond atomName1="N3" atomName2="C2"/>
<Bond atomName1="C2" atomName2="O2"/>
<Bond atomName1="C3'" atomName2="H3'"/>
<Bond atomName1="C3'" atomName2="C2'"/>
<Bond atomName1="C3'" atomName2="O3'"/>
<Bond atomName1="C2'" atomName2="H2'"/>
<Bond atomName1="C2'" atomName2="H2''"/>
<Bond atomName1="O3'" atomName2="HO3'"/>
</Residue> |
|
Ah, I see it now - having drawn things out - I had a 5' phosphate but Amber wanted a 5' hydroxyl group. I've included a PDB for an isolated DT and a short oligo for others who want an example/something easy to start out with. Thanks for the help! |
|
I am also struggling with terminal residue patching when using charmm forcefiled. Could you please comment on the difference between e.g. GLY, GLYN, and GLYC in the charmm36.xml file? Are the "GLYN" and "GLYC" terminal residue capping for GLY? How does the I have Here is my MET residue: |
|
GLY, GLYC, and GLYN are the versions for middle of a chain, C-terminal, and N-terminal respectively. GLY is unusual in this respect. Most amino acids just have one version and use patches for the terminal residues. Your MET residue looks strange. What are the C1, C2, and O1 atoms? Those aren't part of a standard MET residue. |
This comment has been minimized.
This comment has been minimized.
|
Sorry for reopening this issue, but it turns out I still have the patching issue. To make it more clear and simple, how can I add terminal patching to this simple 3 residue peptide? CRYST1 41.604 116.842 91.486 90.00 90.00 90.00 C 2 2 21 8 MODEL 1 ATOM 1 CH3 ACE A 0 35.416 9.009 -18.663 1.00 0.00 C ATOM 2 C ACE A 0 34.218 9.958 -18.693 1.00 0.00 C ATOM 3 O ACE A 0 33.432 9.978 -17.748 1.00 0.00 O ATOM 4 1H ACE A 0 35.403 8.426 -17.715 1.00 0.00 H ATOM 5 2H ACE A 0 35.362 8.313 -19.530 1.00 0.00 H ATOM 6 3H ACE A 0 36.359 9.596 -18.727 1.00 0.00 H ATOM 7 N MET A 1 34.018 10.791 -19.775 1.00 20.33 N ATOM 8 CA MET A 1 32.868 11.699 -19.798 1.00 18.20 C ATOM 9 C MET A 1 31.568 10.891 -19.713 1.00 15.92 C ATOM 10 O MET A 1 31.352 9.991 -20.514 1.00 14.01 O ATOM 11 CB MET A 1 32.903 12.559 -21.061 1.00 21.42 C ATOM 12 CG MET A 1 31.963 13.738 -21.031 1.00 25.49 C ATOM 13 SD MET A 1 32.638 15.208 -21.861 1.00 31.29 S ATOM 14 CE MET A 1 32.598 14.706 -23.443 1.00 28.17 C ATOM 15 H1 MET A 1 34.664 10.775 -20.551 1.00 0.00 H ATOM 16 HA MET A 1 32.926 12.355 -18.930 1.00 0.00 H ATOM 17 HB3 MET A 1 32.676 11.938 -21.927 1.00 0.00 H ATOM 18 HB2 MET A 1 33.920 12.912 -21.229 1.00 0.00 H ATOM 19 HG3 MET A 1 31.724 13.984 -19.996 1.00 0.00 H ATOM 20 HG2 MET A 1 31.018 13.460 -21.499 1.00 0.00 H ATOM 21 HE1 MET A 1 32.982 15.498 -24.086 1.00 0.00 H ATOM 22 HE2 MET A 1 33.215 13.815 -23.558 1.00 0.00 H ATOM 23 HE3 MET A 1 31.571 14.475 -23.726 1.00 0.00 H ATOM 24 N THR A 2 30.712 11.223 -18.741 1.00 9.98 N ATOM 25 CA THR A 2 29.469 10.501 -18.503 1.00 8.85 C ATOM 26 C THR A 2 28.342 11.463 -18.217 1.00 11.00 C ATOM 27 O THR A 2 28.506 12.405 -17.424 1.00 9.24 O ATOM 28 CB THR A 2 29.651 9.536 -17.306 1.00 15.81 C ATOM 29 OG1 THR A 2 30.789 8.703 -17.531 1.00 13.83 O ATOM 30 CG2 THR A 2 28.415 8.664 -17.034 1.00 15.49 C ATOM 31 H THR A 2 30.915 12.004 -18.134 1.00 0.00 H ATOM 32 HA THR A 2 29.222 9.920 -19.391 1.00 0.00 H ATOM 33 HB THR A 2 29.843 10.135 -16.416 1.00 0.00 H ATOM 34 HG1 THR A 2 30.901 8.105 -16.788 1.00 0.00 H ATOM 35 HG21 THR A 2 28.610 8.012 -16.183 1.00 0.00 H ATOM 36 HG22 THR A 2 27.560 9.303 -16.813 1.00 0.00 H ATOM 37 HG23 THR A 2 28.197 8.058 -17.913 1.00 0.00 H ATOM 38 N GLU A 3 27.186 11.203 -18.842 1.00 8.87 N ATOM 39 CA GLU A 3 25.982 11.973 -18.580 1.00 7.86 C ATOM 40 C GLU A 3 25.238 11.354 -17.409 1.00 11.09 C ATOM 41 O GLU A 3 25.183 10.124 -17.284 1.00 10.54 O ATOM 42 CB GLU A 3 25.055 11.978 -19.791 1.00 9.04 C ATOM 43 CG GLU A 3 25.462 12.978 -20.839 1.00 12.16 C ATOM 44 CD GLU A 3 24.509 13.090 -22.010 1.00 19.31 C ATOM 45 OE1 GLU A 3 24.886 13.774 -22.981 1.00 11.19 O ATOM 46 OE2 GLU A 3 23.403 12.500 -21.976 1.00 12.96 O1- ATOM 47 H GLU A 3 27.134 10.454 -19.517 1.00 0.00 H ATOM 48 HA GLU A 3 26.256 12.998 -18.332 1.00 0.00 H ATOM 49 HB3 GLU A 3 24.036 12.188 -19.466 1.00 0.00 H ATOM 50 HB2 GLU A 3 25.030 10.981 -20.232 1.00 0.00 H ATOM 51 HG3 GLU A 3 26.458 12.730 -21.207 1.00 0.00 H ATOM 52 HG2 GLU A 3 25.581 13.958 -20.376 1.00 0.00 H ATOM 53 N NMA A 3A 24.622 12.170 -16.482 1.00 0.00 N ATOM 54 CA NMA A 3A 23.907 11.584 -15.358 1.00 0.00 C ATOM 55 H NMA A 3A 24.667 13.173 -16.584 1.00 0.00 H ATOM 56 1HA NMA A 3A 23.976 10.475 -15.428 1.00 0.00 H ATOM 57 2HA NMA A 3A 24.361 11.929 -14.402 1.00 0.00 H ATOM 58 3HA NMA A 3A 22.839 11.897 -15.386 1.00 0.00 H CONECT 1 4 5 6 CONECT 4 1 CONECT 5 1 CONECT 6 1 ENDMDL END If I use the above PDB file, I get this error: raise ValueError('No template found for residue %d (%s). %s' % (res.index+1, res.name, _findMatchErrors(self, res)))
ValueError: No template found for residue 1 (ACE). The set of atoms is similar to ACET, but it is missing 1 atoms.
Just to be clear, I get the above error only when using |
|
CHARMM handles terminal groups differently from Amber. Amber considers ACE to be a separate residue, while CHARMM treats it as a patch on the existing residue. So change those lines of the PDB file to say it's part of the MET residue. |
|
Unfortunately, that also doesn't work: PDB: CRYST1 41.604 116.842 91.486 90.00 90.00 90.00 C 2 2 21 8 MODEL 1 ATOM 1 CH3 MET A 1 35.416 9.009 -18.663 1.00 0.00 C ATOM 2 C MET A 1 34.218 9.958 -18.693 1.00 0.00 C ATOM 3 O MET A 1 33.432 9.978 -17.748 1.00 0.00 O ATOM 4 1H MET A 1 35.403 8.426 -17.715 1.00 0.00 H ATOM 5 2H MET A 1 35.362 8.313 -19.530 1.00 0.00 H ATOM 6 3H MET A 1 36.359 9.596 -18.727 1.00 0.00 H ATOM 7 N MET A 1 34.018 10.791 -19.775 1.00 20.33 N ATOM 8 CA MET A 1 32.868 11.699 -19.798 1.00 18.20 C ATOM 9 C MET A 1 31.568 10.891 -19.713 1.00 15.92 C ATOM 10 O MET A 1 31.352 9.991 -20.514 1.00 14.01 O ATOM 11 CB MET A 1 32.903 12.559 -21.061 1.00 21.42 C ATOM 12 CG MET A 1 31.963 13.738 -21.031 1.00 25.49 C ATOM 13 SD MET A 1 32.638 15.208 -21.861 1.00 31.29 S ATOM 14 CE MET A 1 32.598 14.706 -23.443 1.00 28.17 C ATOM 15 H1 MET A 1 34.664 10.775 -20.551 1.00 0.00 H ATOM 16 HA MET A 1 32.926 12.355 -18.930 1.00 0.00 H ATOM 17 HB3 MET A 1 32.676 11.938 -21.927 1.00 0.00 H ATOM 18 HB2 MET A 1 33.920 12.912 -21.229 1.00 0.00 H ATOM 19 HG3 MET A 1 31.724 13.984 -19.996 1.00 0.00 H ATOM 20 HG2 MET A 1 31.018 13.460 -21.499 1.00 0.00 H ATOM 21 HE1 MET A 1 32.982 15.498 -24.086 1.00 0.00 H ATOM 22 HE2 MET A 1 33.215 13.815 -23.558 1.00 0.00 H ATOM 23 HE3 MET A 1 31.571 14.475 -23.726 1.00 0.00 H ATOM 24 N THR A 2 30.712 11.223 -18.741 1.00 9.98 N ATOM 25 CA THR A 2 29.469 10.501 -18.503 1.00 8.85 C ATOM 26 C THR A 2 28.342 11.463 -18.217 1.00 11.00 C ATOM 27 O THR A 2 28.506 12.405 -17.424 1.00 9.24 O ATOM 28 CB THR A 2 29.651 9.536 -17.306 1.00 15.81 C ATOM 29 OG1 THR A 2 30.789 8.703 -17.531 1.00 13.83 O ATOM 30 CG2 THR A 2 28.415 8.664 -17.034 1.00 15.49 C ATOM 31 H THR A 2 30.915 12.004 -18.134 1.00 0.00 H ATOM 32 HA THR A 2 29.222 9.920 -19.391 1.00 0.00 H ATOM 33 HB THR A 2 29.843 10.135 -16.416 1.00 0.00 H ATOM 34 HG1 THR A 2 30.901 8.105 -16.788 1.00 0.00 H ATOM 35 HG21 THR A 2 28.610 8.012 -16.183 1.00 0.00 H ATOM 36 HG22 THR A 2 27.560 9.303 -16.813 1.00 0.00 H ATOM 37 HG23 THR A 2 28.197 8.058 -17.913 1.00 0.00 H ATOM 38 N GLU A 3 27.186 11.203 -18.842 1.00 8.87 N ATOM 39 CA GLU A 3 25.982 11.973 -18.580 1.00 7.86 C ATOM 40 C GLU A 3 25.238 11.354 -17.409 1.00 11.09 C ATOM 41 O GLU A 3 25.183 10.124 -17.284 1.00 10.54 O ATOM 42 CB GLU A 3 25.055 11.978 -19.791 1.00 9.04 C ATOM 43 CG GLU A 3 25.462 12.978 -20.839 1.00 12.16 C ATOM 44 CD GLU A 3 24.509 13.090 -22.010 1.00 19.31 C ATOM 45 OE1 GLU A 3 24.886 13.774 -22.981 1.00 11.19 O ATOM 46 OE2 GLU A 3 23.403 12.500 -21.976 1.00 12.96 O1- ATOM 47 H GLU A 3 27.134 10.454 -19.517 1.00 0.00 H ATOM 48 HA GLU A 3 26.256 12.998 -18.332 1.00 0.00 H ATOM 49 HB3 GLU A 3 24.036 12.188 -19.466 1.00 0.00 H ATOM 50 HB2 GLU A 3 25.030 10.981 -20.232 1.00 0.00 H ATOM 51 HG3 GLU A 3 26.458 12.730 -21.207 1.00 0.00 H ATOM 52 HG2 GLU A 3 25.581 13.958 -20.376 1.00 0.00 H ATOM 53 N NMA A 3A 24.622 12.170 -16.482 1.00 0.00 N ATOM 54 CA NMA A 3A 23.907 11.584 -15.358 1.00 0.00 C ATOM 55 H NMA A 3A 24.667 13.173 -16.584 1.00 0.00 H ATOM 56 1HA NMA A 3A 23.976 10.475 -15.428 1.00 0.00 H ATOM 57 2HA NMA A 3A 24.361 11.929 -14.402 1.00 0.00 H ATOM 58 3HA NMA A 3A 22.839 11.897 -15.386 1.00 0.00 H CONECT 1 4 5 6 CONECT 4 1 CONECT 5 1 CONECT 6 1 ENDMDL END the code: And the entire output: building protein and solvating...
/home/asgharrazavi/miniconda3/lib/python3.7/site-packages/simtk/openmm/app/internal/pdbstructure.py:537: UserWarning: WARNING: duplicate atom (ATOM 9 C MET A 1 31.568 10.891 -19.713 1.00 15.92 C , ATOM 2 C MET A 1 34.218 9.958 -18.693 1.00 0.00 C )
warnings.warn("WARNING: duplicate atom (%s, %s)" % (atom, old_atom._pdb_string(old_atom.serial_number, atom.alternate_location_indicator)))
/home/asgharrazavi/miniconda3/lib/python3.7/site-packages/simtk/openmm/app/internal/pdbstructure.py:537: UserWarning: WARNING: duplicate atom (ATOM 10 O MET A 1 31.352 9.991 -20.514 1.00 14.01 O , ATOM 3 O MET A 1 33.432 9.978 -17.748 1.00 0.00 O )
warnings.warn("WARNING: duplicate atom (%s, %s)" % (atom, old_atom._pdb_string(old_atom.serial_number, atom.alternate_location_indicator)))
Adding hydrogens...
unmatched residues: [, ]
Traceback (most recent call last):
File "00_simple.py", line 14, in
modeller.addHydrogens(omm_forcefield)
File "/home/asgharrazavi/miniconda3/lib/python3.7/site-packages/simtk/openmm/app/modeller.py", line 943, in addHydrogens
system = forcefield.createSystem(newTopology, rigidWater=False, nonbondedMethod=CutoffNonPeriodic)
File "/home/asgharrazavi/miniconda3/lib/python3.7/site-packages/simtk/openmm/app/forcefield.py", line 1146, in createSystem
raise ValueError('No template found for residue %d (%s). %s' % (res.index+1, res.name, _findMatchErrors(self, res)))
ValueError: No template found for residue 1 (MET). The set of atoms is similar to AOBT, but it is missing 2 atoms.
|
|
It appears that part of the problem might be in detecting hydrogen atoms. In the above PDB file, the first residue (capped MET) has 12 hydrogens, but the program detects 11. # of hydrogens for residue : ['H', 'H2', 'H3', 'HA', 'HB2', 'HB3', 'HE1', 'HE2', 'HE3', 'HG2', 'HG3'] 11 # of hydrogens for residue : ['H', 'HA', 'HB', 'HG1', 'HG21', 'HG22', 'HG23'] 7 # of hydrogens for residue : ['H', 'HA', 'HB2', 'HB3', 'HG2', 'HG3'] 6 # of hydrogens for residue : ['H', 'H1', 'H2', 'H3'] 4 To get above information, I added the code hydrogens = [h for h in spec.hydrogens if (variant is None and pH <= h.maxph) or (h.variants is None and pH <= h.maxph) or (h.variants is not None and variant in h.variants)]
hydrogens = [h for h in hydrogens if h.terminal is None or (isNTerminal and h.terminal == 'N') or (isCTerminal and h.terminal == 'C')]
hydrogens = [h for h in hydrogens if h.parent in parentNames]
print('# of hydrogens for residue %s:' %(residue), [i5.name for i5 in hydrogens], len(hydrogens)) #ASGHAR
# Loop over atoms in the residue, adding them to the new topology along with required hydrogens.
for parent in residue.atoms():
|
|
I think I see. Because the PDB reader expects ACE to be a separate residue, and doesn't add bonds correctly if it's merged. But CHARMM expects it to be part of the same residue, and doesn't work properly if it's separate. Sigh. Things would be simpler if everyone in the community would just agree on a consistent way of treating things like this. CHARMM is the nonstandard one in this case. The PDB lists ACE as a separate residue, and most programs follow it. But CHARMM calls it a patch instead of a residue, which requires it to be merged. Anyone have suggestions on a clean way of dealing with this? |
|
Tagging in @j-wags, since we're working on the Open Force Field Our approach will be more in line with perceiving biopolymer residues using industry-standard SMARTS matches, and then making this information available for traditional template-based schemes. |
|
For the moment, is there a way to manually give information to topology builder for the caped N- and C-terminal residues? There is only 2 of them, so, it will help a lot just to have a manual adjustment before the problem is fundamentally fixed. Thanks. |
|
You can add the missing bonds to the Topology by calling |
|
I created an N-capped MET residue template like this: omm_forcefield = app.ForceField('charmm36.xml', 'charmm36/water.xml')
met = omm_forcefield._templates['MET']
metn = copy.deepcopy(met)
atoms = [atom for atom in met.atoms]
cay = copy.deepcopy(atoms[0])
cy = copy.deepcopy(atoms[0])
oy = copy.deepcopy(atoms[0])
hy1 = copy.deepcopy(atoms[0])
hy2 = copy.deepcopy(atoms[0])
hy3 = copy.deepcopy(atoms[0])
hy1.parameters, hy1.name, hy1.type, = {'charge':0.09 } ,"HY1" ,"HA3",
hy2.parameters, hy2.name, hy2.type, = {'charge':0.09 } ,"HY2" ,"HA3",
hy3.parameters, hy3.name, hy3.type, = {'charge':0.09 } ,"HY3" ,"HA3",
cy.parameters, cy.name, cy.type, = {'charge':0.51 } ,"CY" ,"C",
oy.parameters, oy.name, oy.type, = {'charge':-0.51} ,"OY" ,"O",
metn.addAtom(cay) #index 17
metn.addAtom(hy1) # 18
metn.addAtom(hy2) # 19
metn.addAtom(hy3) # 20
metn.addAtom(cy) # 21
metn.addAtom(oy) # 22
metn.addBond(17, 18)
metn.addBond(17, 19)
metn.addBond(17, 20)
metn.addBond(17, 21)
metn.addBond(21, 22)
metn.addBond(21, 0)
omm_forcefield._templates['METN'] = metn
I am pretty sure all the extra bonds are added now, but still get the "no template found" error :( |
|
Did you add the missing bonds to the topology like I suggested? |
This comment has been minimized.
This comment has been minimized.
This comment has been minimized.
This comment has been minimized.
|
Oh, I think I just fixed it! As the program outputs as "warnings" when reading PDB file, there are duplicate names for some heavy atoms in the last residue (GLU). I changed the last residue patching atom name like this and now it is working without error. ATOM 38 N GLU A 3 27.186 11.203 -18.842 1.00 8.87 N ATOM 39 CA GLU A 3 25.982 11.973 -18.580 1.00 7.86 C ATOM 40 C GLU A 3 25.238 11.354 -17.409 1.00 11.09 C ATOM 41 O GLU A 3 25.183 10.124 -17.284 1.00 10.54 O ATOM 42 CB GLU A 3 25.055 11.978 -19.791 1.00 9.04 C ATOM 43 CG GLU A 3 25.462 12.978 -20.839 1.00 12.16 C ATOM 44 CD GLU A 3 24.509 13.090 -22.010 1.00 19.31 C ATOM 45 OE1 GLU A 3 24.886 13.774 -22.981 1.00 11.19 O ATOM 46 OE2 GLU A 3 23.403 12.500 -21.976 1.00 12.96 O1- ATOM 47 H GLU A 3 27.134 10.454 -19.517 1.00 0.00 H ATOM 48 HA GLU A 3 26.256 12.998 -18.332 1.00 0.00 H ATOM 49 HB3 GLU A 3 24.036 12.188 -19.466 1.00 0.00 H ATOM 50 HB2 GLU A 3 25.030 10.981 -20.232 1.00 0.00 H ATOM 51 HG3 GLU A 3 26.458 12.730 -21.207 1.00 0.00 H ATOM 52 HG2 GLU A 3 25.581 13.958 -20.376 1.00 0.00 H ATOM 53 NT GLU A 3 24.622 12.170 -16.482 1.00 0.00 N ATOM 54 CAT GLU A 3 23.907 11.584 -15.358 1.00 0.00 C ATOM 55 HT GLU A 3 24.667 13.173 -16.584 1.00 0.00 H ATOM 56 1HA GLU A 3 23.976 10.475 -15.428 1.00 0.00 H ATOM 57 2HA GLU A 3 24.361 11.929 -14.402 1.00 0.00 H ATOM 58 3HA GLU A 3 22.839 11.897 -15.386 1.00 0.00 H So, the lesson is not to ignore "warnings" :). EDIT: I also needed to change hydrogen atoms in first residue like this: ATOM 4 1HA MET A 1 35.403 8.426 -17.715 1.00 0.00 H ATOM 5 2HA MET A 1 35.362 8.313 -19.530 1.00 0.00 H ATOM 6 3HA MET A 1 36.359 9.596 -18.727 1.00 0.00 H Having trouble with un-hiding some previous comments, so here is the final PDB file and the code:working PDB:CRYST1 41.604 116.842 91.486 90.00 90.00 90.00 C 2 2 21 8 MODEL 1 ATOM 1 CY3 MET A 1 35.416 9.009 -18.663 1.00 0.00 C ATOM 2 CY MET A 1 34.218 9.958 -18.693 1.00 0.00 C ATOM 3 OY MET A 1 33.432 9.978 -17.748 1.00 0.00 O ATOM 4 1HA MET A 1 35.403 8.426 -17.715 1.00 0.00 H ATOM 5 2HA MET A 1 35.362 8.313 -19.530 1.00 0.00 H ATOM 6 3HA MET A 1 36.359 9.596 -18.727 1.00 0.00 H ATOM 7 N MET A 1 34.018 10.791 -19.775 1.00 20.33 N ATOM 8 CA MET A 1 32.868 11.699 -19.798 1.00 18.20 C ATOM 9 C MET A 1 31.568 10.891 -19.713 1.00 15.92 C ATOM 10 O MET A 1 31.352 9.991 -20.514 1.00 14.01 O ATOM 11 CB MET A 1 32.903 12.559 -21.061 1.00 21.42 C ATOM 12 CG MET A 1 31.963 13.738 -21.031 1.00 25.49 C ATOM 13 SD MET A 1 32.638 15.208 -21.861 1.00 31.29 S ATOM 14 CE MET A 1 32.598 14.706 -23.443 1.00 28.17 C ATOM 15 HN MET A 1 34.664 10.775 -20.551 1.00 0.00 H ATOM 16 HA MET A 1 32.926 12.355 -18.930 1.00 0.00 H ATOM 17 HB3 MET A 1 32.676 11.938 -21.927 1.00 0.00 H ATOM 18 HB2 MET A 1 33.920 12.912 -21.229 1.00 0.00 H ATOM 19 HG3 MET A 1 31.724 13.984 -19.996 1.00 0.00 H ATOM 20 HG2 MET A 1 31.018 13.460 -21.499 1.00 0.00 H ATOM 21 HE1 MET A 1 32.982 15.498 -24.086 1.00 0.00 H ATOM 22 HE2 MET A 1 33.215 13.815 -23.558 1.00 0.00 H ATOM 23 HE3 MET A 1 31.571 14.475 -23.726 1.00 0.00 H ATOM 24 N THR A 2 30.712 11.223 -18.741 1.00 9.98 N ATOM 25 CA THR A 2 29.469 10.501 -18.503 1.00 8.85 C ATOM 26 C THR A 2 28.342 11.463 -18.217 1.00 11.00 C ATOM 27 O THR A 2 28.506 12.405 -17.424 1.00 9.24 O ATOM 28 CB THR A 2 29.651 9.536 -17.306 1.00 15.81 C ATOM 29 OG1 THR A 2 30.789 8.703 -17.531 1.00 13.83 O ATOM 30 CG2 THR A 2 28.415 8.664 -17.034 1.00 15.49 C ATOM 31 H THR A 2 30.915 12.004 -18.134 1.00 0.00 H ATOM 32 HA THR A 2 29.222 9.920 -19.391 1.00 0.00 H ATOM 33 HB THR A 2 29.843 10.135 -16.416 1.00 0.00 H ATOM 34 HG1 THR A 2 30.901 8.105 -16.788 1.00 0.00 H ATOM 35 HG21 THR A 2 28.610 8.012 -16.183 1.00 0.00 H ATOM 36 HG22 THR A 2 27.560 9.303 -16.813 1.00 0.00 H ATOM 37 HG23 THR A 2 28.197 8.058 -17.913 1.00 0.00 H ATOM 38 N GLU A 3 27.186 11.203 -18.842 1.00 8.87 N ATOM 39 CA GLU A 3 25.982 11.973 -18.580 1.00 7.86 C ATOM 40 C GLU A 3 25.238 11.354 -17.409 1.00 11.09 C ATOM 41 O GLU A 3 25.183 10.124 -17.284 1.00 10.54 O ATOM 42 CB GLU A 3 25.055 11.978 -19.791 1.00 9.04 C ATOM 43 CG GLU A 3 25.462 12.978 -20.839 1.00 12.16 C ATOM 44 CD GLU A 3 24.509 13.090 -22.010 1.00 19.31 C ATOM 45 OE1 GLU A 3 24.886 13.774 -22.981 1.00 11.19 O ATOM 46 OE2 GLU A 3 23.403 12.500 -21.976 1.00 12.96 O1- ATOM 47 H GLU A 3 27.134 10.454 -19.517 1.00 0.00 H ATOM 48 HA GLU A 3 26.256 12.998 -18.332 1.00 0.00 H ATOM 49 HB3 GLU A 3 24.036 12.188 -19.466 1.00 0.00 H ATOM 50 HB2 GLU A 3 25.030 10.981 -20.232 1.00 0.00 H ATOM 51 HG3 GLU A 3 26.458 12.730 -21.207 1.00 0.00 H ATOM 52 HG2 GLU A 3 25.581 13.958 -20.376 1.00 0.00 H ATOM 53 NT GLU A 3 24.622 12.170 -16.482 1.00 0.00 N ATOM 54 CAT GLU A 3 23.907 11.584 -15.358 1.00 0.00 C ATOM 55 HT GLU A 3 24.667 13.173 -16.584 1.00 0.00 H ATOM 56 1HA GLU A 3 23.976 10.475 -15.428 1.00 0.00 H ATOM 57 2HA GLU A 3 24.361 11.929 -14.402 1.00 0.00 H ATOM 58 3HA GLU A 3 22.839 11.897 -15.386 1.00 0.00 H ENDMDL END Code:from simtk.openmm.app import PDBFile
from simtk.openmm import app
print('building protein and solvating...')
# Load the force field parameters through OpenMM.
omm_forcefield = app.ForceField('charmm36.xml')
# Load the PDB file.
prot_pdb_file = PDBFile('test.pdb')
modeller = app.Modeller(prot_pdb_file.topology, prot_pdb_file.positions)
top = prot_pdb_file.topology
## ADDING EXTRA BONDS ##
# Adding extra bonds to the first residue
print('Total number of bonds before adding extra bonds:', top.bonds)
res = [res for res in top.residues()][0]
atoms = [atom for atom in res.atoms()]
bonds = [bond for bond in res.bonds()]
print('Bonds for residue 1 before adding extra bonds to topology:')
for bond in bonds:
print('%s %s' %(bond[0],bond[1]))
top.addBond(atoms[0],atoms[3])
top.addBond(atoms[0],atoms[4])
top.addBond(atoms[0],atoms[5])
top.addBond(atoms[0],atoms[1])
top.addBond(atoms[1],atoms[2])
top.addBond(atoms[1],atoms[6])
res = [res for res in top.residues()][0]
bonds = [bond for bond in res.bonds()]
print('Bonds for residue 1 after adding extra bonds to topology:')
for bond in bonds:
print('%s %s' %(bond[0],bond[1]))
# Adding extra bonds to the last residue
print('Total number of bonds before adding extra bonds to last residue:', top.bonds)
res = [res for res in top.residues()][-1]
atoms = [atom for atom in top.atoms()]
bonds = [bond for bond in res.bonds()]
print('Bonds for the last residue before adding extra bonds to topology:')
for bond in bonds:
print('%s %s' %(bond[0],bond[1]))
top.addBond(atoms[39],atoms[52])
top.addBond(atoms[52],atoms[53])
top.addBond(atoms[52],atoms[54])
top.addBond(atoms[53],atoms[55])
top.addBond(atoms[53],atoms[56])
top.addBond(atoms[53],atoms[57])
res = [res for res in top.residues()][-1]
bonds = [bond for bond in res.bonds()]
print('Bonds for the last residue after adding extra bonds to topology:')
for bond in bonds:
print('%s %s' %(bond[0],bond[1]))
print('Total number of bonds after adding extra bonds:', top.bonds)
## FINISHED ADDING EXTRA BONDS ##
print('Adding hydrogens...')
modeller.addHydrogens(omm_forcefield)
|
|
Hi, Thank you for the solution! I followed this method, but my code still didn't work. My system is Ac-Glu-NMe-Ca2+. Here is my pdb file: My code is: There are strange bonds appeared: Do I need to build a new template of capped Glu? |
|
It recognizes the atom names "HT1", "HT2", and "HT3" as synonyms for "H", "H2", and "H3". And since those atoms would be bonded to N in a normal residue, it adds those bonds. Try giving them different names. @asgharrazavi called them "1HA", "2HA", and "3HA" in the example above. |
|
It worked! Thank you! |
|
I think there is still an issue here. When I try to write the final PDB file using a suggestion from here, I see that there are extra hydrogen atoms: code to write PDB: with open('output.pdb', 'w') as f:
PDBFile.writeFile(modeller.topology, modeller.positions, f)
output PDB for the first residue: REMARK 1 CREATED WITH OPENMM 7.4.1, 2020-09-30 CRYST1 41.604 116.842 91.486 90.00 90.00 90.00 P 1 1 ATOM 1 CY3 MET A 1 35.416 9.009 -18.663 1.00 0.00 C ATOM 2 CY MET A 1 34.218 9.958 -18.693 1.00 0.00 C ATOM 3 OY MET A 1 33.432 9.978 -17.748 1.00 0.00 O ATOM 4 1HY MET A 1 35.394 8.440 -17.706 1.00 0.00 H ATOM 5 2HY MET A 1 35.363 8.279 -19.498 1.00 0.00 H ATOM 6 3HY MET A 1 36.373 9.568 -18.712 1.00 0.00 H ATOM 7 N MET A 1 34.018 10.791 -19.775 1.00 0.00 N ATOM 8 H2 MET A 1 33.930 10.074 -20.521 1.00 0.00 H ATOM 9 H3 MET A 1 33.937 10.265 -18.847 1.00 0.00 H ATOM 10 CA MET A 1 32.868 11.699 -19.798 1.00 0.00 C ATOM 11 C MET A 1 31.568 10.891 -19.713 1.00 0.00 C ATOM 12 O MET A 1 31.352 9.991 -20.514 1.00 0.00 O ATOM 13 CB MET A 1 32.903 12.559 -21.061 1.00 0.00 C ATOM 14 CG MET A 1 31.963 13.738 -21.031 1.00 0.00 C ATOM 15 SD MET A 1 32.638 15.208 -21.861 1.00 0.00 S ATOM 16 CE MET A 1 32.598 14.706 -23.443 1.00 0.00 C ATOM 17 H MET A 1 34.933 11.267 -19.802 1.00 0.00 H ATOM 18 HA MET A 1 32.921 12.342 -18.927 1.00 0.00 H ATOM 19 HB3 MET A 1 32.698 11.950 -21.974 1.00 0.00 H ATOM 20 HB2 MET A 1 33.943 12.956 -21.175 1.00 0.00 H ATOM 21 HG3 MET A 1 31.754 14.026 -19.976 1.00 0.00 H ATOM 22 HG2 MET A 1 30.979 13.460 -21.477 1.00 0.00 H ATOM 23 HE1 MET A 1 32.887 15.535 -24.124 1.00 0.00 H ATOM 24 HE2 MET A 1 33.297 13.863 -23.624 1.00 0.00 H ATOM 25 HE3 MET A 1 31.577 14.377 -23.733 1.00 0.00 H These are the extra hydrogens: ATOM 8 H2 MET A 1 33.930 10.074 -20.521 1.00 0.00 H ATOM 9 H3 MET A 1 33.937 10.265 -18.847 1.00 0.00 H I tried renaming H atoms for N-terminal patching for different names, but wasn't successful. Strangely, the patched last residue is fine. |
|
I deleted the extra atoms using PDB: CRYST1 41.604 116.842 91.486 90.00 90.00 90.00 C 2 2 21 8 MODEL 1 ATOM 1 CY3 MET A 1 35.416 9.009 -18.663 1.00 0.00 C ATOM 2 CY MET A 1 34.218 9.958 -18.693 1.00 0.00 C ATOM 3 OY MET A 1 33.432 9.978 -17.748 1.00 0.00 O ATOM 4 1HY MET A 1 35.403 8.426 -17.715 1.00 0.00 H ATOM 5 2HY MET A 1 35.362 8.313 -19.530 1.00 0.00 H ATOM 6 3HY MET A 1 36.359 9.596 -18.727 1.00 0.00 H ATOM 7 N MET A 1 34.018 10.791 -19.775 1.00 20.33 N ATOM 8 CA MET A 1 32.868 11.699 -19.798 1.00 18.20 C ATOM 9 C MET A 1 31.568 10.891 -19.713 1.00 15.92 C ATOM 10 O MET A 1 31.352 9.991 -20.514 1.00 14.01 O ATOM 11 CB MET A 1 32.903 12.559 -21.061 1.00 21.42 C ATOM 12 CG MET A 1 31.963 13.738 -21.031 1.00 25.49 C ATOM 13 SD MET A 1 32.638 15.208 -21.861 1.00 31.29 S ATOM 14 CE MET A 1 32.598 14.706 -23.443 1.00 28.17 C ATOM 15 HN MET A 1 34.664 10.775 -20.551 1.00 0.00 H ATOM 16 HA MET A 1 32.926 12.355 -18.930 1.00 0.00 H ATOM 17 HB3 MET A 1 32.676 11.938 -21.927 1.00 0.00 H ATOM 18 HB2 MET A 1 33.920 12.912 -21.229 1.00 0.00 H ATOM 19 HG3 MET A 1 31.724 13.984 -19.996 1.00 0.00 H ATOM 20 HG2 MET A 1 31.018 13.460 -21.499 1.00 0.00 H ATOM 21 HE1 MET A 1 32.982 15.498 -24.086 1.00 0.00 H ATOM 22 HE2 MET A 1 33.215 13.815 -23.558 1.00 0.00 H ATOM 23 HE3 MET A 1 31.571 14.475 -23.726 1.00 0.00 H ATOM 24 N THR A 2 30.712 11.223 -18.741 1.00 9.98 N ATOM 25 CA THR A 2 29.469 10.501 -18.503 1.00 8.85 C ATOM 26 C THR A 2 28.342 11.463 -18.217 1.00 11.00 C ATOM 27 O THR A 2 28.506 12.405 -17.424 1.00 9.24 O ATOM 28 CB THR A 2 29.651 9.536 -17.306 1.00 15.81 C ATOM 29 OG1 THR A 2 30.789 8.703 -17.531 1.00 13.83 O ATOM 30 CG2 THR A 2 28.415 8.664 -17.034 1.00 15.49 C ATOM 31 H THR A 2 30.915 12.004 -18.134 1.00 0.00 H ATOM 32 HA THR A 2 29.222 9.920 -19.391 1.00 0.00 H ATOM 33 HB THR A 2 29.843 10.135 -16.416 1.00 0.00 H ATOM 34 HG1 THR A 2 30.901 8.105 -16.788 1.00 0.00 H ATOM 35 HG21 THR A 2 28.610 8.012 -16.183 1.00 0.00 H ATOM 36 HG22 THR A 2 27.560 9.303 -16.813 1.00 0.00 H ATOM 37 HG23 THR A 2 28.197 8.058 -17.913 1.00 0.00 H ATOM 38 N GLU A 3 27.186 11.203 -18.842 1.00 8.87 N ATOM 39 CA GLU A 3 25.982 11.973 -18.580 1.00 7.86 C ATOM 40 C GLU A 3 25.238 11.354 -17.409 1.00 11.09 C ATOM 41 O GLU A 3 25.183 10.124 -17.284 1.00 10.54 O ATOM 42 CB GLU A 3 25.055 11.978 -19.791 1.00 9.04 C ATOM 43 CG GLU A 3 25.462 12.978 -20.839 1.00 12.16 C ATOM 44 CD GLU A 3 24.509 13.090 -22.010 1.00 19.31 C ATOM 45 OE1 GLU A 3 24.886 13.774 -22.981 1.00 11.19 O ATOM 46 OE2 GLU A 3 23.403 12.500 -21.976 1.00 12.96 O1- ATOM 47 H GLU A 3 27.134 10.454 -19.517 1.00 0.00 H ATOM 48 HA GLU A 3 26.256 12.998 -18.332 1.00 0.00 H ATOM 49 HB3 GLU A 3 24.036 12.188 -19.466 1.00 0.00 H ATOM 50 HB2 GLU A 3 25.030 10.981 -20.232 1.00 0.00 H ATOM 51 HG3 GLU A 3 26.458 12.730 -21.207 1.00 0.00 H ATOM 52 HG2 GLU A 3 25.581 13.958 -20.376 1.00 0.00 H ATOM 53 NT GLU A 3 24.622 12.170 -16.482 1.00 0.00 N ATOM 54 CAT GLU A 3 23.907 11.584 -15.358 1.00 0.00 C ATOM 55 HT GLU A 3 24.667 13.173 -16.584 1.00 0.00 H ATOM 56 1HA GLU A 3 23.976 10.475 -15.428 1.00 0.00 H ATOM 57 2HA GLU A 3 24.361 11.929 -14.402 1.00 0.00 H ATOM 58 3HA GLU A 3 22.839 11.897 -15.386 1.00 0.00 H ENDMDL END Code: from simtk.openmm.app import PDBFile
from simtk.openmm import app
from simtk import unit
from simtk.openmm import Platform
from simtk.openmm import LangevinIntegrator
print('building protein and solvating...')
# Load the force field parameters through OpenMM.
omm_forcefield = app.ForceField('charmm36.xml','charmm36/water.xml')
# Load the PDB file.
prot_pdb_file = PDBFile('test.pdb')
modeller = app.Modeller(prot_pdb_file.topology, prot_pdb_file.positions)
top = prot_pdb_file.topology
## ADDING EXTRA BONDS ##
# Adding extra bonds to the first residue
res = [res for res in top.residues()][0]
atoms = [atom for atom in res.atoms()]
bonds = [bond for bond in res.bonds()]
top.addBond(atoms[0],atoms[3])
top.addBond(atoms[0],atoms[4])
top.addBond(atoms[0],atoms[5])
top.addBond(atoms[0],atoms[1])
top.addBond(atoms[1],atoms[2])
top.addBond(atoms[1],atoms[6])
res = [res for res in top.residues()][0]
bonds = [bond for bond in res.bonds()]
# Adding extra bonds to the last residue
res = [res for res in top.residues()][-1]
atoms = [atom for atom in top.atoms()]
bonds = [bond for bond in res.bonds()]
top.addBond(atoms[39],atoms[52])
top.addBond(atoms[52],atoms[53])
top.addBond(atoms[52],atoms[54])
top.addBond(atoms[53],atoms[55])
top.addBond(atoms[53],atoms[56])
top.addBond(atoms[53],atoms[57])
res = [res for res in top.residues()][-1]
bonds = [bond for bond in res.bonds()]
## FINISHED ADDING EXTRA BONDS ##
print('Adding hydrogens...')
modeller.addHydrogens(omm_forcefield)
# Deleting exra hydrogens
h2 = [atom for atom in modeller.topology.atoms() if atom.name == 'H2' and atom.residue.name == 'MET']
print('atom to be deleted:', h2)
modeller.delete(h2)
h3 = [atom for atom in modeller.topology.atoms() if atom.name == 'H3' and atom.residue.name == 'MET']
print('atom to be deleted:', h3)
modeller.delete(h3)
print('Adding solvent...')
# here we can also add ionic concentration (read addSolvent?)
modeller.addSolvent(omm_forcefield, model='tip3p', padding=1*unit.nanometer, ionicStrength=0.1*unit.molar, positiveIon='Na+', negativeIon='Cl-')
with open('output.pdb', 'w') as f:
PDBFile.writeFile(modeller.topology, modeller.positions, f)
prot_system = omm_forcefield.createSystem(modeller.topology, nonbondedMethod=app.PME, rigidWater=True)
## simulations
dt = 0.005*unit.picoseconds
temperature = 310*unit.kelvin
friction = 1.0/unit.picosecond
constraintTolerance = 0.000001
log_name = 'log.log'
# Simulation Options
steps = 160000
equilibrationSteps = 0
pdbReporter = app.PDBReporter('test.pdb', 20)
dataReporter = app.StateDataReporter(log_name, 1, totalSteps=steps, step=True, time=True, speed=True, progress=True, elapsedTime=True, remainingTime=True, potentialEnergy=True, kineticEnergy=True, totalEnergy=True, temperature=True, volume=True, density=True, separator=',')
integrator = LangevinIntegrator(temperature, friction, dt)
integrator.setConstraintTolerance(constraintTolerance)
simulation = app.Simulation(modeller.topology, prot_system, integrator)
simulation.context.setPositions(modeller.positions)
simulation.reporters.append(pdbReporter)
simulation.reporters.append(dataReporter)
simulation.minimizeEnergy()
print('writing minimized structure...')
positions = simulation.context.getState(getPositions=True).getPositions()
PDBFile.writeFile(simulation.topology, positions, open('minimized.pdb', 'w'))
# Set velocity and loadCheckpoint if available
simulation.context.setVelocitiesToTemperature(temperature)
simulation.currentStep = 0
# Simulate
print('Simulating...')
simulation.step(steps)
Output: building protein and solvating... Adding hydrogens... before applying patches: [template, matches]: [None, None] after applying patches: [template, matches]: [None, None] atom to be deleted: [<Atom 7 (H2) of chain 0 residue 0 (MET)>] Adding solvent...
writing minimized structure...
Simulating...
Traceback (most recent call last):
File "final_code_on_git_edit2.py", line 96, in
simulation.step(steps)
File "/home/asgharrazavi/miniconda3/lib/python3.7/site-packages/simtk/openmm/app/simulation.py", line 132, in step
self._simulate(endStep=self.currentStep+steps)
File "/home/asgharrazavi/miniconda3/lib/python3.7/site-packages/simtk/openmm/app/simulation.py", line 235, in _simulate
self._generate_reports(unwrapped, False)
File "/home/asgharrazavi/miniconda3/lib/python3.7/site-packages/simtk/openmm/app/simulation.py", line 254, in _generate_reports
reporter.report(self, state)
File "/home/asgharrazavi/miniconda3/lib/python3.7/site-packages/simtk/openmm/app/statedatareporter.py", line 195, in report
self._checkForErrors(simulation, state)
File "/home/asgharrazavi/miniconda3/lib/python3.7/site-packages/simtk/openmm/app/statedatareporter.py", line 347, in _checkForErrors
raise ValueError('Energy is NaN')
ValueError: Energy is NaN
I wonder if I am adding solvent properly? |
|
So, the problem was my MD time step. I was inappropriately using 5 fs for simulation time step without hydrogen mass repartitioning. It works with 2 fs time step :) |
|
I am trying to simulate a 2 amino acid sequence using amber force field in openmm. But there is always an error `from simtk.openmm.app import * pdb = PDBFile('Leu-Phe.pdb') i have also attached the pdb file.
Can you comment what is the problem and how to resolve it. @peastman |

|
What order are they supposed to be in? The PHE is listed before the LEU, but the reside numbers are the other way around: PHE is residue 2 and LEU is residue 1. And the PHE is terminated (notice the OXT and HXT atoms), as if it were at the end of a chain, while the LEU is not terminated. |
|
I created new pdb file of only Phe residue and tried to simulate that using openmm but still there is error coming. |
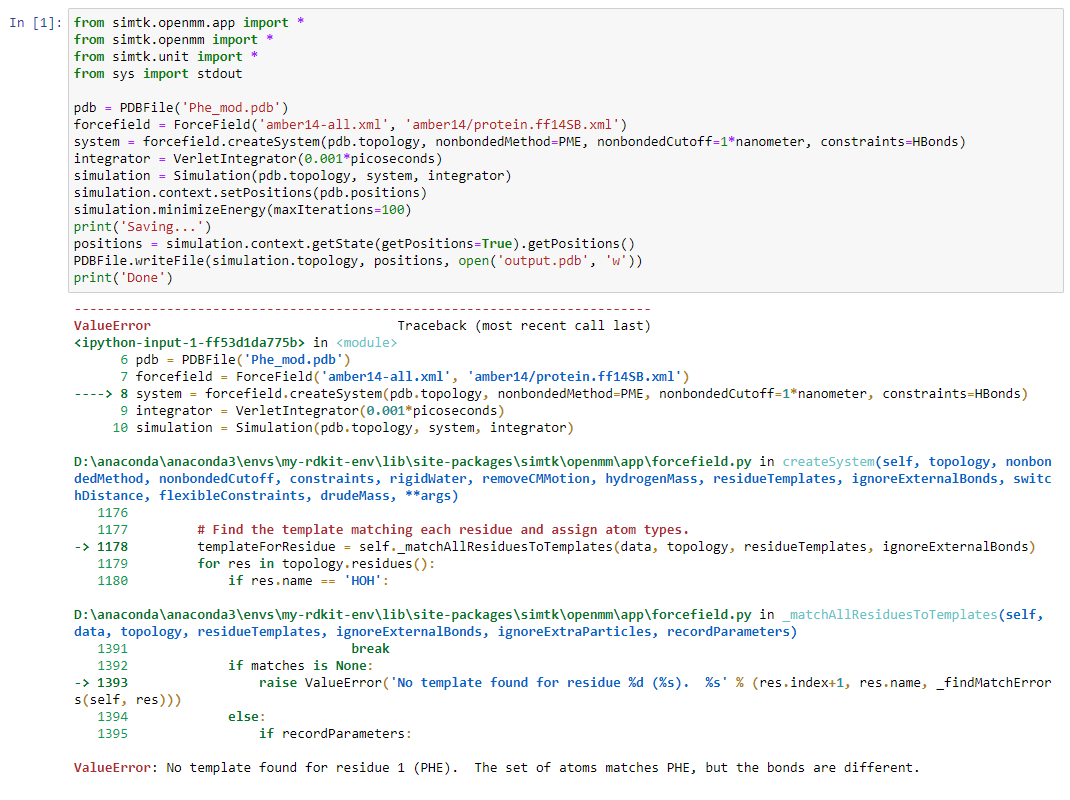

|
Could you attach a ZIPped version of the PDB file to your issue? From a quick inspection of the image of the PDB file contents, it looks like the N and C terminus have just been truncated---natural amino acids end in zwitterionic termini, with NH3(+)- at the N-terminus and -CO2(-) at the C-terminus. You only have one proton at the N-terminus and one oxygen at the C-terminus, suggesting you clipped out an internal PHE residue and left dangling bonds. OpenMM won't know how to match that. |
|
The first one is a normal Phe amino acid with the terminals, the second one we have removed the OH from the C terminal and H from the N terminal. Please let me know how to fix the pdb files. |

|
Try using OpenMM Setup. It can fix improperly terminated chains. |
|
Thanks for the reply. I have been also using the openmm setup but the same error is coming. |
|
Can you attach a peptide pdb file which can be simulated in openmm, Because i have been trying different amino acid sequence and every time there is some error. |
|
Thank you @peastman , i am able to simulate ala-ala-ala pdb file, i was getting error due to my pdb file, I want to know how to create the correct pdb file from a given amino acid sequence. Is there any script to do that. Because the way i am creating the pdb file i am not able to simulate it, but the connections are all right as i can see from its 3d visualization. |


|
Your file is severely malformed. All the atoms for each residue need to go together. Your file begins by listing all the non-hydrogens for all residues, then follows with the hydrogens for all residues. RDKit just views your SMILES string as an arbitrary molecule, not realizing it's made up of residues whose atoms need to be kept together. I don't know what tools people use for building peptides from scratch. It isn't something I've had to do much. Maybe someone else can suggest something. |
|
Thanks for replying. there is a function in rdkit MolFromSequence which generates correct mol format of the amino acid sequence which is only for peptides. |
Hi! I'm trying to go a little bit farther than the getting started example and try out the same thing with a short oligonucleotide instead of a protein. However, I keep getting the same error of "No template found for residue."
I thought that this might be an issue with my PDB formation, so I tried a few oligos from the PDB (6GN4 and 6GPI) both of which have the same issue for me. I also created two new "oligos" - one which is single-stranded DNA and another which is just dTMP. Both give the same issue.
For the dTMP this is the error I get:
which seems to be "recognising" the thymidine but still isn't able to go forward?
I'm sure I'm missing something small, but any help would be appreciated.
This is my code, almost exactly the same as the sample code:
The text was updated successfully, but these errors were encountered: