Version 0.1.1, written in Python 3.6.6
Takes fastq files from 10x single cell RNA sequencing, clusters cells using Seurat, and can be used to produce read count matrices in a cluster level. You can also quantify reads per cluster having predefined clusters.
TrusTEr depends on several external software. We provide a Docker container and a conda environment for a quick-start.
TrusTEr requires:
- Cellranger
- R (version 3.6)
- Seurat
- TEtranscripts
- STAR aligner
- subset-bam and bamtofastq from 10x Genomics
- Velocyto
The package has been tested in Unix systems only and supports only SLURM job submissions.
If you fulfill the requirements, you can install via pip:
pip install truster
As single cell technologies haven't developed to the point where we can get the needed sequencing depth to study transposable elements expression, trusTEr seeks produce more reliable results by combining reads from closely related cells to gain information on a cell type level.
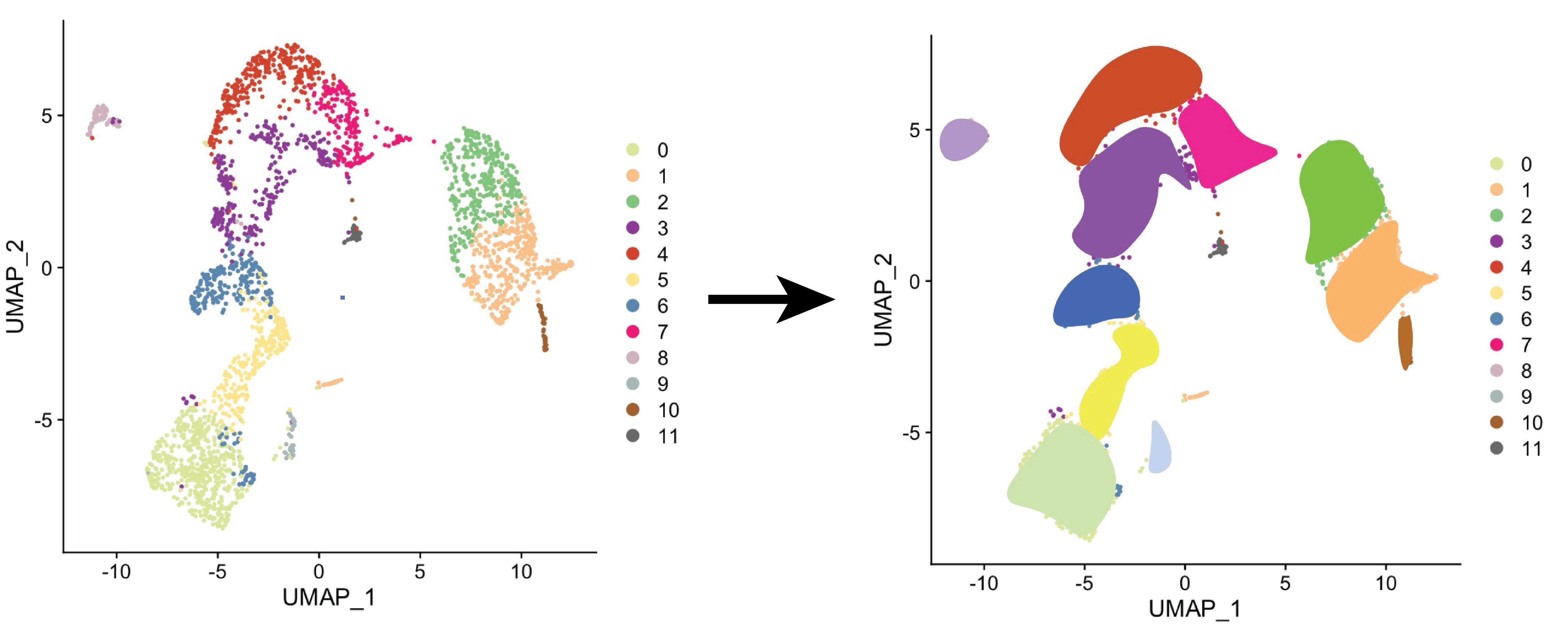
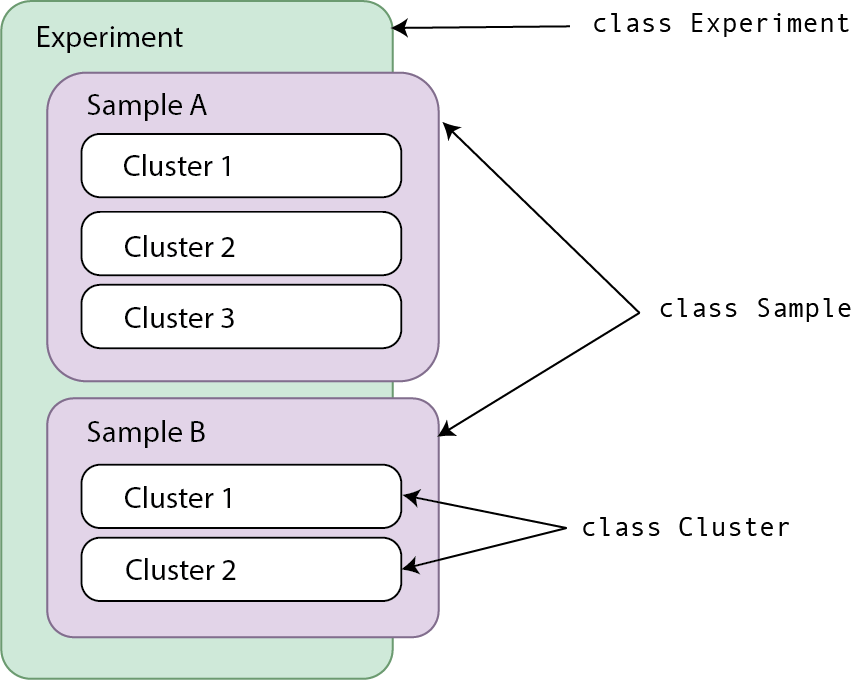
trusTEr uses composition assiciation to relate three main classes:
- Experiment: Includes information about the experiment as a whole. This is the main object you will be working with.
- Name is required, description is optional.
- Register samples by providing a path to its fastq files.
- Sample: Created by giving a path to fastq files
- Name and ID required.
- Cluster: Created by running
get_clusters()ormerge_samples()functions (Orset_clusters_outdir()orset_merge_samples_outdir()).
This package is meant to be run with the following workflow:
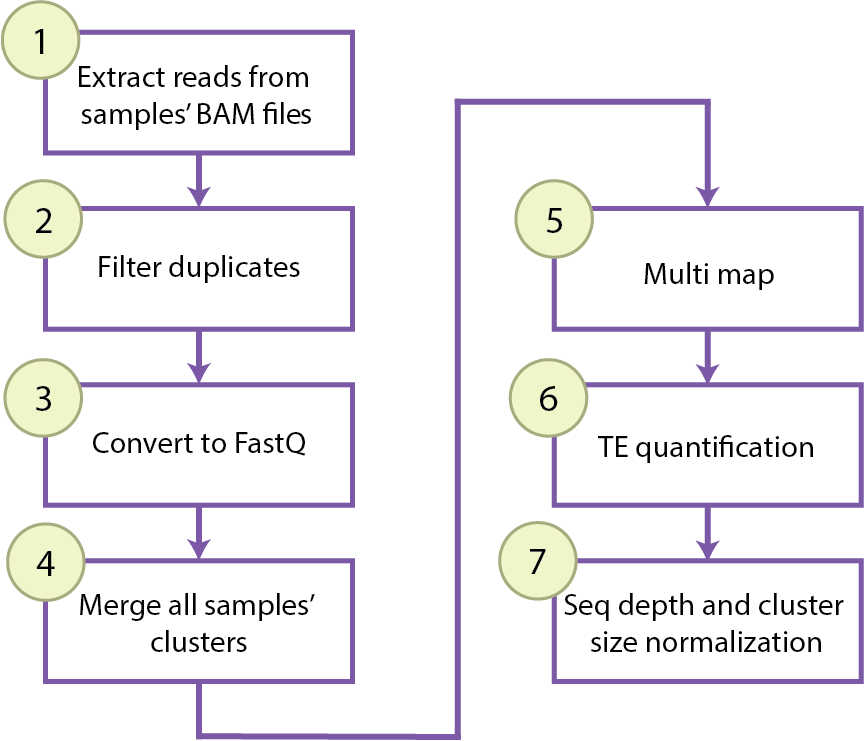
Depending on the object type, you have access to different functions to go through these steps.
Experiment is the main object one would work with. Here you will register_sample() or register_samples_from_path().
An object of type Sample has access to step 7 and some handy wrappers to use cellRanger, perform clustering with Seurat and run and plot RNA velocity. These functions can be called for all registered samples from your object of type Experiment (See quantify(), get_clusters_all_samples(), velocity_all_samples(), plot_velocity_all_samples()).
The need for the class Experiment is clearer once the user wants to merge samples (See merge_samples()) or to run the same workflow for all the samples' clusters.
The user won't work directly with an object of type Cluster, but this class includes all the functions needed to go through steps 1-6 of the workflow. Instead of running this pipeline individually for each cluster, one can run the workflow for each cluster of each registered sample using the Experiment function process_clusters(mode = "per_sample", ...).
One can also partition the workflow and run step by step in all registered samples or in a combined clustering using the transitioning functions of Experiment to call the needed functions in the class Cluster (See tsv_to_bam_clusters(), filter_UMIs_clusters(), bam_to_fastq_clusters(), concatenate_lanes_clusters(), merge_clusters(), map_clusters(), TE_counts_clusters(), normalize_TE_counts()).
At the moment of construction of an Experiment object, you can declare slurm_path and a modules_path. These files will be checked whenever a function can be run as an sbatch job.
For the moment, trusTEr only works with slurm systems. You need to create two json files:
1. slurmPath Declaring the sbatch options per function. You need to declare a field per function listed that you will use:
- quantify
- get_clusters
- merge_samples
- tsv_to_bam
- filter_UMIs
- bam_to_fastq
- map_cluster
- TE_count
- velocity
- plot_velocity
As a brief example:
{
"__default__" :
{
"account" : "myaccount",
"time" : "00:15:00",
"nodes" : 1,
"partition" : "dell",
"output" : "%j.out",
"error" : "%j.err",
"job-name" : "%j"
},
"quantify" :
{
"account" : "myaccount",
"time" : "10:00:00",
"nodes" : 1,
"tasks-per-node" : 20,
"partition" : "dell",
"output" : "%j.quantify.out",
"error" : "%j.quantify.err",
"job-name" : "%j.quantify"
},
"get_clusters" : {...},
"merge_samples" : {...},
"tsv_to_bam" : {...},
"filter_UMIs" : {...},
"bam_to_fastq" : {...},
"map_cluster" : {...},
"TE_count" : {...},
"velocity" : {...},
"plot_velocity" : {...}
}
2. modulesPath
In a slurm system, many times you need to load modules for a software to be available. In this json file you declare the name of the needed modules. You need to declare a field per function listed that you will use:
- quantify
- get_clusters
- merge_samples
- tsv_to_bam
- filter_UMIs
- bam_to_fastq
- map_cluster
- TE_count
- TE_count_unique
- velocity
- normalize_TE_counts
- plot_TE_expression
As an example:
{
"quantify":["cellranger/3.1.0"],
"velocity":["GCC/7.3.0-2.30", "SAMtools/1.9", "velocyto/0.17.17"],
"get_clusters":["GCC/9.3.0", "OpenMPI/4.0.3", "Seurat/3.1.5-R-4.0.0"],
"merge_samples":["GCC/9.3.0", "OpenMPI/4.0.3", "Seurat/3.1.5-R-4.0.0"],
"tsv_to_bam":["subset-bam/1.0"],
"filter_UMIs":["GCC/7.3.0-2.30", "OpenMPI/3.1.1", "Pysam/0.15.1-Python-3.6.6"],
"bam_to_fastq":["bamtofastq/1.2.0"],
"map_cluster":["GCC/5.4.0-2.26", "OpenMPI/1.10.3", "STAR/2.6.0c"],
"TE_count":["icc/2018.1.163-GCC-6.4.0-2.28", "OpenMPI/2.1.2", "TEToolkit/2.0.3-Python-2.7.14"],
"TE_count_unique": ["GCC/7.3.0-2.30", "OpenMPI/3.1.1", "Subread/1.6.3"],
"normalize_TE_counts":["GCC/9.3.0", "OpenMPI/4.0.3", "Seurat/3.1.5-R-4.0.0"],
"plot_TE_expression":["GCC/9.3.0", "OpenMPI/4.0.3", "Seurat/3.1.5-R-4.0.0"]
}
If you don't need to load any modules for a software to be available, you can leave the respective list empty.
1: Sbatch signaling error 2: The command ran failed 3: Incorrect input 4: File not found True: False:
You can read the functions' documentation and some tutorials at https://raquelgarza.github.io/truster/