Can't Embed a molecule #2996
Comments
|
@MherMatevosyan |
|
@MherMatevosyan |
|
@iiitmjay, thank you for your suggestion, but I need to use the energy minimized version in other calculations, so I would like to understand if those molecules are even possible to embed and have their energy minimized. And if it is not possible to do, I would like to understand the underlying reason for that. |
|
@greglandrum Sir, if you have time then please have a look on this conversation. |
|
@MherMatevosyan sorry for the slow reply on this one. Finishing up the most recent RDKit release has kept me overly busy. As you're seeing, when When this fails, I generally then take a close look at the molecule. Here's what you have: That includes this substructure, which I believe is the source of the problem: I think it's going to be difficult to find a 3D structure of that ring system that is even close to being physically reasonable. Because the RDKit's conformer generator uses rules that attempt to generate physically reasonable structures, they fail here. We might be able to figure out how to get a conformer for things like this, but I want to check first: do you believe that these are physically reasonable molecules? |


|
@greglandrum is there a simpler version of 3d conformation that checks for viability of fragment structures based on some rules like it is done in the sanitizer to avoid these failures? |
|
@denfromufa I imagine that it's possible to put together a set of heuristics to recognize substructures which may be problematic, but the RDKit doesn't have anything like that built in. |
|
Just wanted to chime in - I recently ran into a similar problem and I found this thread from Google. The molecule is an oligosaccharide and the SMILES string is:
I'm also attaching a screenshot of the notebook cells where I saw this problem. I went up to 100,000 attempts and still no luck. The GLYCAM carbohydrate builder has no problem with building a structure, I've attached that as well. Maybe that's the solution I should've tried in the beginning, but my sense was that RDKit can routinely embed molecules bigger than this one. |

|
@leeping - what happened after you followed the advice given in the error message and added explicit hydrogen atoms? |
|
@leeping , as @jasondbiggs implied: you should be adding Hs to molecules before attempting to embed them. It would be good to know if you still see the problem then |
|
Hi Jason and Greg, Thanks for the advice - calling AddHs() before attempting to embed resolved the issue for that molecule. My apologies for not checking. That molecule is actually a truncated version of the original that also included two lipid tails. I had previously confirmed that AddHs() did not resolve the issue for the larger molecule, but I neglected to check it for the truncated version. If the failure for the larger molecule is of interest, I'm including the screenshot and SMILES string here: (This is with RDKit version 2022.03.5).
|
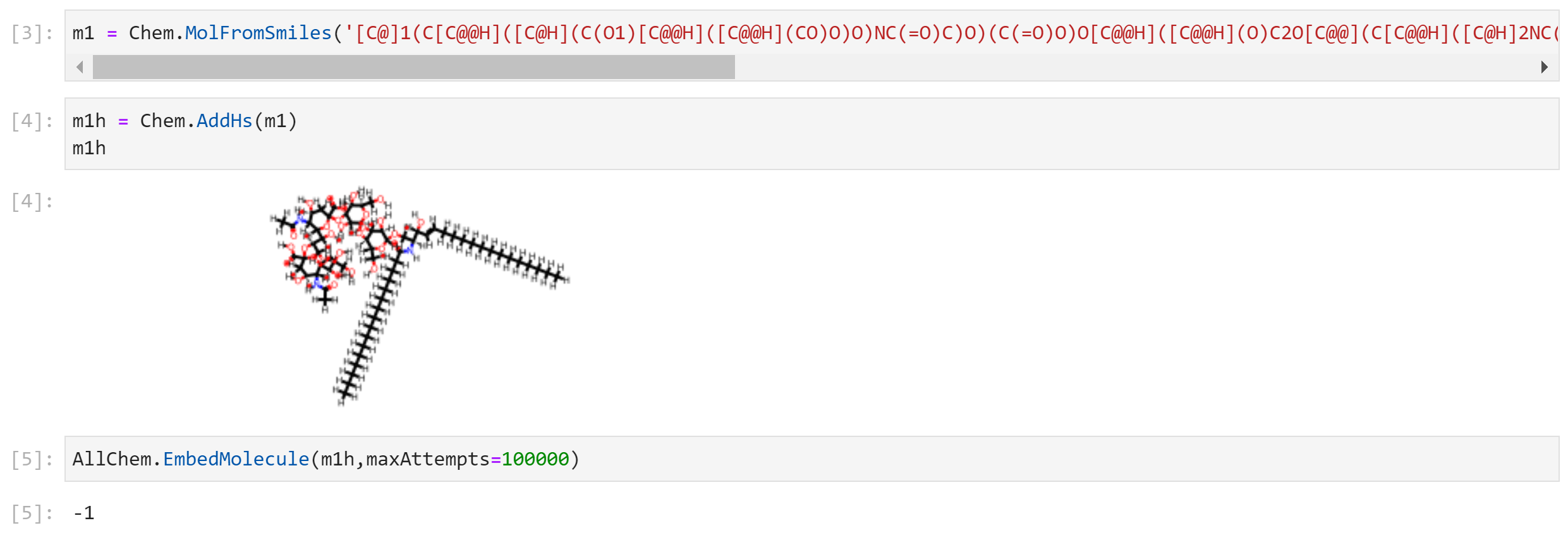
|
One more example case where rdkit fails at embedding which is less extreme than the above molecules. Surprisingly if you remove the hydrogens it works (so if you run the EmbedMolecule on m1 instead of m1h) and you can add the hydrogens back in afterwards but I assume that's not very correct. from rdkit import Chem
from rdkit.Chem import AllChem
m1 = Chem.MolFromSmiles('C1CC2CCC21')
m1h = Chem.AddHs(m1)
AllChem.EmbedMolecule(m1h, maxAttempts=100000)
|

Description:
It seems rdkit can't embed some molecules. Tried with
enforceChirality = False,ignoreSmoothingFailures = False. Those didn't work. Could you help, please?Example SMILES:
C=CC1=C(N)Oc2cc1c(-c1cc(C(C)O)cc(=O)cc1C1NCC(=O)N1)c(OC)c2OC
C=CC1=C(N)Oc2cc1c(C1=CC(C(O)CN)=CC(O)C=C1C1NCC(=O)N1)c(OC)c2OC
The text was updated successfully, but these errors were encountered: