Welcome to CeleScope Discussions! #27
Replies: 6 comments 18 replies
-
|
Hi, I use Telescope to analyze single cell RNA data generated by Singleron. -- celescope.tools.step.get_slot_key - WARNING - Assay not found in sample_summary.metrics i think its related with the --sample Attribute. thanks! |
Beta Was this translation helpful? Give feedback.
-
|
You need to run |

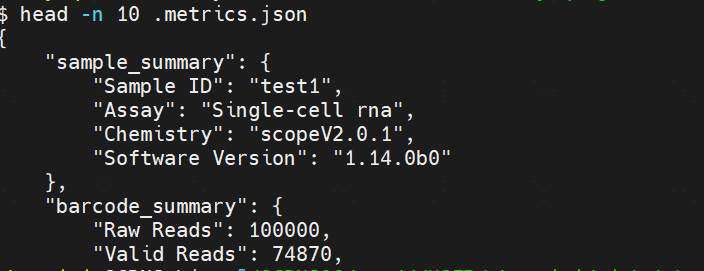
Beta Was this translation helpful? Give feedback.
-
|
Hi, I'm kind of new to bioinformatics and I wanted to try out CeleScope according to the user guide. |
Beta Was this translation helpful? Give feedback.
-
|
CeleScope only runs on Linux systems. |
Beta Was this translation helpful? Give feedback.
-
|
Hi, How to install it on public HPC system, with no conda command available? |
Beta Was this translation helpful? Give feedback.
-
|
Thanks for that. Maenwhile, Wondering if you know something about this error that I encountered with standard installation (mamba;pip) on my desktop after pip installation command; |
Beta Was this translation helpful? Give feedback.
-
|
This error message is incomplete and does not show the specific error. Please attach the command and the full log. |
Beta Was this translation helpful? Give feedback.
-
|
Full log `pip install celescope × Getting requirements to build wheel did not run successfully. note: This error originates from a subprocess, and is likely not a problem with pip. × Getting requirements to build wheel did not run successfully. note: This error originates from a subprocess, and is likely not a problem with pip. |
Beta Was this translation helpful? Give feedback.
-
|
It may be related to the gcc you were using. velocyto-team/velocyto.py#88 I think it can be fixed by installing pysam with mamba instead of pip |
Beta Was this translation helpful? Give feedback.
-
|
Thanks, seems |
Beta Was this translation helpful? Give feedback.
-
|
Hi, I was looking for information about index files, I got for each sample in context to scRNAseq. Do I need to provide this as an input somewhere? I could not find this info here https://github.com/singleron-RD/CeleScope/blob/master/doc/assay/multi_rna.md |
Beta Was this translation helpful? Give feedback.
-
|
If your data is already demultiplexed (you have separate R1.fastq and R2.fastq files for each sample), then you don't really need it. |
Beta Was this translation helpful? Give feedback.
-
|
Great Got you. Thanks |
Beta Was this translation helpful? Give feedback.
-
|
Where can I find the complete documentation for Celescope? I've received the final report, which includes two folders in the output—'raw' and 'filtered.' I'm unsure about which input to use for Seurat (raw or filtered). I couldn't find any documentation that explains the output details comprehensively, including the default filters applied. Additionally, I'm curious about the tool or method Celescope uses for clustering cell types. When I use filtered data as input in Seurat, I observe different clusters than those generated by Celescope. Can you provide insights into this discrepancy?" |
Beta Was this translation helpful? Give feedback.
-
|
The filter matrix is used for downstream analysis: CeleScope uses Scanpy to do the downstream analysis which may use different paramaters as what you use in Seurat. For example, the default resolution used in CeleScope(Scanpy) is 1.2. |
Beta Was this translation helpful? Give feedback.
-
|
Thanks |
Beta Was this translation helpful? Give feedback.
-
|
I am running some DynaSCOPE samples and I wanted to get familiar with the celescope analysis. I first downloaded and installed the conda packages like on this site Then I downloaded all the data and scripts like this Then I generated the mouse genome like this: Then I modified arguments in the dynaseq script to my mouse genomeDir path like this Then I ran the test like this Sadly this is the point where I get an error message. I looked at the log file (attached 1) and it seems to be a problem that it cannot find the mt_gene_list and gtf files. However, when I go to my genomeDIR I do think that I see these files (attached 2) Do you know how I can fix this problem? I have limited bioinformatics experience, so it is difficult for me to logically describe my problem to you. I hope you understand my question. |

Beta Was this translation helpful? Give feedback.
-
|
Thanks for the quick reply! The celescope_genome.config does indeed not include that data: [files] [meta] |
Beta Was this translation helpful? Give feedback.
-
|
I tried it again from scratch and now it works! :) |
Beta Was this translation helpful? Give feedback.
-
|
Good to know. The previous |
Beta Was this translation helpful? Give feedback.
-
|
Hi! I hope you don't mind, but I have another question. I tried to run the pipeline now with my own data. I sequenced my DynaSCOPE libraries wich looked good on bioanalyzer and concentration. Multiple samples were pooled on the sequencing flowcell. So afterwards I demultiplexed the data with the adater sequences from the DynaSCOPE manual (I used cellranger which is uses on the bcl2fastq method from illumina). |
Beta Was this translation helpful? Give feedback.
-
|
DynaSCOPE requires at least 72bp in R1 read to work. Below is the library structure: |
Beta Was this translation helpful? Give feedback.
-
👋 Welcome!
We’re using Discussions as a place to connect with other members of our community. We hope that you:
build together 💪.
To get started, comment below with an introduction of yourself and tell us about what you do with this community.
Beta Was this translation helpful? Give feedback.
All reactions