As a novel analysis tool developed for quality control and preprocessing of FASTQ and SAM/BAM data, SOAPnuke includes 5 modules for different usage scenarios, namely filter, filterHts, filterStLFR, filtersRNA and filterMeta.
filter: Preprocess FASTQ files, include trimming (adapter, low quality end and etc.) if set, discarding (adapter, low quality, high N base ratio and etc.) and generating statistic report.
filterHts: Preprocess BAM/CRAM files. The process procedure remains the same as filter module.
(Note: Input BAM/CRAM files should be sorted by readID when it contains Paired-End data.)
filterStLFR: Preprocess stLFR FASTQ files, added with a barcode-detection step at the beginning, and support FASTQ files list as input.
filtersRNA: Preprocess sRNA FASTQ files. Since it is still under testing, please inform us if you encounter any bug.
filterMeta: Preprocess Meta FASTQ files. Since it is still under testing, please inform us if you encounter any bug.
SOAPnuke 2.X version shows an excellent performance compared with 1.X version. An great acceleration has been accomplished by refactoring the whole framework, optimizing multithreading and IO.
This table presents a benchmark result on 628M Paired-End 150bp reads. As thread number increases, user time obviously decreases.
| Software | ThreadNum | RunTime(min) | MaxMem(MB) | Parameter |
|---|---|---|---|---|
| SOAPnuke | 16 | 35.7 | 2270 | filter module |
| SOAPnuke | 8 | 48.4 | 881 | filter module |
| SOAPnuke | 4 | 72.1 | 275 | filter module |
| fastp | 8 | 62.0 | 1004 | -A -w 8 |
gcc: 4.7 or higher
zlib: 1.2.3.5 or higher
htslib: 1.9 or higher
pthread library
git clone https://github.com/BGI-flexlab/SOAPnuke.git
cd SOAPnuke
// Considering rarely been used and complex compile dependency, we turn off filterHts module by default.
// If you want to use filterHts module, please set USEHTS true in Makefile like this:
// USEHTS=true
make
All usages start with executable file SOAPnuke, and different modules are invoked with different sub-commands. Here are some usage examples:
#filter:
#QC the input fastq and extract 10M clean reads to the output files.
echo "totalReadsNum=10000000" >config.txt
SOAPnuke filter -1 test.r1.fq.gz -2 test.r2.fq.gz -C clean_1.fq.gz -D clean_2.fq.gz -o result -T 8 -c config.txt
#filterHts:
SOAPnuke filterHts --ref chr21.fa -1 input.bam -2 output.cram -o result
SOAPnuke filterHts -1 input.bam -2 output.bam -o result
#filterStLFR:
filterStLFR -1 fq1.list -2 fq2.list -C clean1.gz -D clean2.gz -o result -T 8 -c configIf set trim-related parameters(no trim if not set), do trimming first:
Read ID
If parameter “index” set in config file, remove index sequence from read ID.
Once “index” is set, if seqType is 0(default value), read ID would be expected like:
@FCD1PB1ACXX:4:1101:1799:2201#GAAGCACG/2,
“#GAAGCACG” would be removed then.
If seqType is 1, read ID would be expected like:
@HISEQ:310:C5MH9ANXX:1:1101:3517:20432:N:0:TCGGTCAC,
“:TCGGTCAC” would be removed then.
Read sequence and quality
First, the cutting length of all trimming type would be calculated, including hard trim, low quality end trim, adapter trim and tail-polyG trim. The longest cutting would be performed.
- hard trim: directly remove a certain length sequence from head or tail on read sequence
- low quality end trim: remove low quality base starting from end until quality higher than cutoff
- adapter trim: when adapter was found, the base sequence and quality sequence would be trimmed from the start position which match adapter
- tail-polyG trim: if polyG number is greater than cutoff, then these polyG sequence in tail would be trimmed
Then do filtering:
Note that the read pair would be both discarded both when any of which fails to pass QC.
Priority(High to Low):
- Tile, may be used in some types of BGI data.
If you want to discard reads with certain tile ID, set the parameter like “1101-1104,1205”.
- Fov, may be used in data from zebra-platform.
If you want to discard reads with certain FOV ID, set the parameter like “C001R003,C003R004”.
- Minimal read length
Discard a read with sequence length shorter than the parameter.
- Maximal read length
Discard a read with sequence length longer than the parameter.
- N ratio
Discard a read with N base ratio not smaller than the parameter.
- High A ratio
Discard a read with A base ratio not smaller than the parameter.
- polyX number (X means any one base)
Discard a read with poly-X number not smaller than the parameter.
- Low quality base ratio
Discard a read with low-quality bases ratio not smaller than the parameter.
- Mean quality
Discard a read of which mean quality of sequence smaller than the parameter.
- Overlapped length if PE
Discard a read pair which is suspected to be overlapped longer then the parameter.
- Adapter
Discard a read which contains an adapter.
- -1 / --fq1
fq1 file(required), .gz or normal text format are both supported
- -2 / --fq2
fq2 file(used when process PE data), format should be same as fq1 file, both are gz or both are normal text
- -C / --cleanFq1
reads which passed QC from fq1 file would output to this file
- -D / --cleanFq2
reads which passed QC from fq2 file would output to this file
- -o / --out
Output directory. Processed fq files and statistical results would be output to here
- -f / --adapter1
adapter sequence or list file of read1
- -r / --adapter2
adapter sequence or list file of read2
- -J / --ada_trim
trim read when find adapter, it’s a bool parameter, default is false which means discard the read when find adapter
- -T / --thread
threads number used in process, default value is 6
- -c / --configFile
config file which include uncommonly used parameters. Each line contains a parameter, e.g., for value needed parameter: adaMis=2, for bool parameter: contam_trim, which means set mode as discard when find contaminant sequence
- -l / --lowQual
low quality threshold, default value is 5
- -q / --qualRate
low quality rate threshold, default value is 0.5
- -n / --nRate
N rate threshold, default value is 0.05
- -m / --mean
low average quality threshold, if you want discard reads with low average quality, you can set a value. The software do NOT check this item by default
- -p / --highA
ratio of A threshold in a read, the software do NOT check this item by default
- -g / --polyG_tail
polyG number threshold in read tail, the software do NOT check this item by default
- -X / --polyX
polyX number threshold, the software do NOT check this item by default
- -4 / --minReadLen
read minimal length, default value is 30
- -h / --help
Show help information
- -v / --version
Show version information
Here we only present options different from filter module.
- -E / --ref
reference file(required when process cram format)
- -1
input bam/cram file(required)
- -2
output bam/cram file(required)
Here we only present options different from filter module.
- -1 / --fq1
Support FASTQ files list as input
- -2 / --fq2
Support FASTQ files list as input
- ctMatchR
Contaminant sequence shortest consistent matching ratio [default:0.2]
- seqType
Sequence fq name type, 0->old fastq name, 1->new fastq name [0]
old fastq name: @FCD1PB1ACXX:4:1101:1799:2201#GAAGCACG/2
new fastq name: @HISEQ:310:C5MH9ANXX:1:1101:3517:2043 2:N:0:TCGGTCAC
- trimFq1
trim fq1 file name(gz format) [optional]
- trimFq2
trim fq2 file name [optional]. If trim related parameters were set on, these output files would include the total reads which only do trimming. For example, if read A failed QC after trimming, it will still output to -R/-W, but not to -C/-D
- tile
tile number to ignore reads, such as [1101-1104,1205]
- fov
fov number to ignore reads (only for zebra-platform data), such as [C001R003,C003R004]
- barcodeListPath
barcode list of two columns:sequence and barcodeID
- barcodeRegionStr
barcode regions, such as: 101_10,117_10,145_10 or 101_10,117_10,133_10
- notCutNoLFR
do not cut sequence when fail found barcode
- inputAsList
input file list not a file
- tenX
output tenX format
- outFileType
output file format: fastq or fasta[default: fastq]
- index
remove index
- totalReadsNum
number/fraction of reads you want to keep in the output clean FASTQ file(cannot be assigned when -w is given). It will extract reads randomly through the total clean FASTQ file by default, you also can get the head reads for save time by add head suffix to the integer
- trim
trim some bp of the read's head and tail, they means: (PE type:read1's head and tail and read2's head and tail [0,0,0,0]; SE type:read head and tail [0,0])
- trimBadHead
Trim from head ends until meeting high-quality base or reach the length threshold, set (quality threshold,MaxLengthForTrim) [0,0]
- trimBadTail
Trim from tail ends until meeting high-quality base or reach the length threshold, set (quality threshold,MaxLengthForTrim) [0,0]
- overlap
filter the small insert size.Not filter until the value exceed 1 [-1]
- mis
the maximum mismatch ratio when find overlap between PE reads(depend on -O) [0.1]
- patch
reads number of a patch processed [400000]
- qualSys
quality system 1:64, 2:33 [default:2]
- outQualSys
out quality system 1:64, 2:33 [default:2]
- maxReadLen
read max length, default 49 for filtersRNA, the software do NOT check this item by default in other modules
- cleanOutSplit
max reads number in each output clean FASTQ file
- pe_info
Add /1, /2 at the end of FASTQ name. [default: not add]
- baseConvert
convert base when write data, example: TtoU , means convert base T to base U in the output
- log
log file output path
The three scripts in src/Rscripts/ are used for plotting QC stats from SOAPnuke.
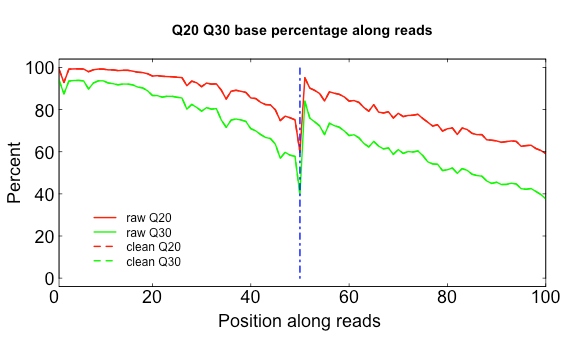
Q20Q30.R
USAGE:
Rscript src/Rscripts/Q20Q30.R Distribution_of_Q20_Q30_bases_by_read_position_1.txt Distribution_of_Q20_Q30_bases_by_read_position_2.txt q2030.png
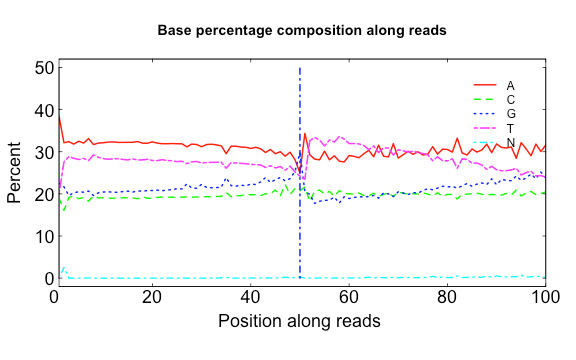
base.R
USAGE:
Rscript src/Rscripts/base.R Base_distributions_by_read_position_1.txt Base_distributions_by_read_position_2.txt raw.png clean.png
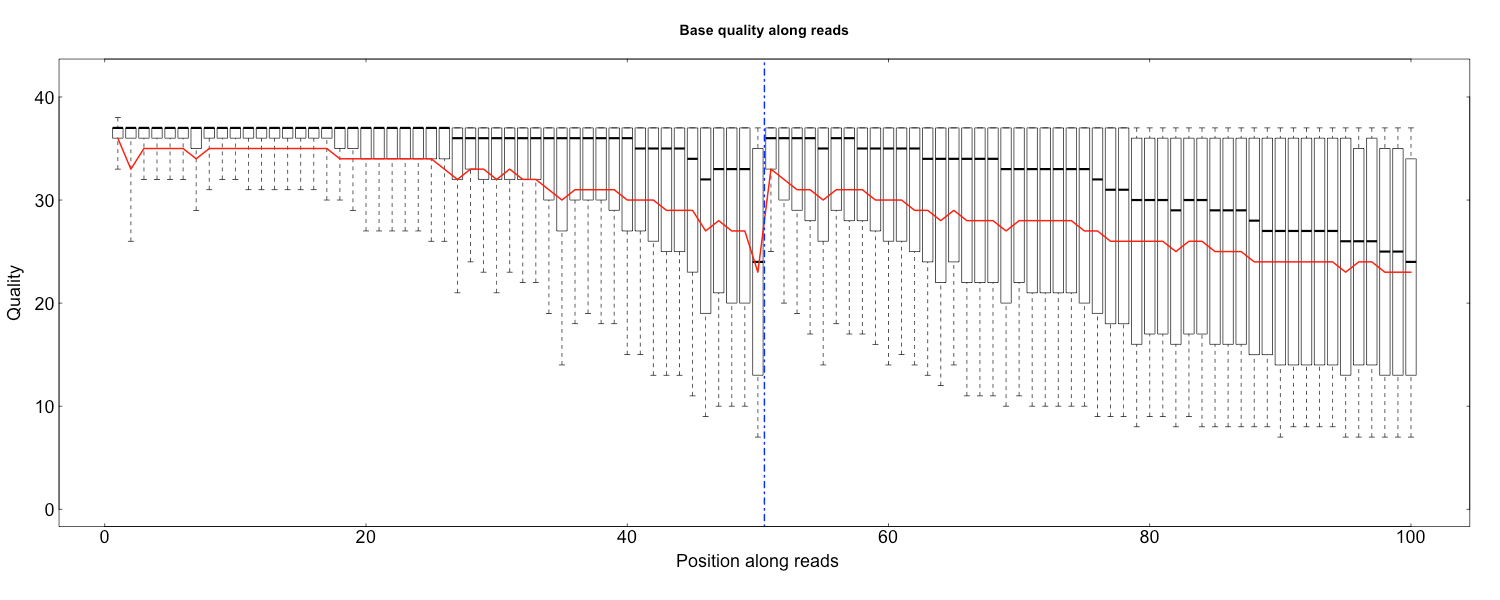
quality.R
Rscript src/Rscripts/quality.R Base_quality_value_distribution_by_read_position_1.txt Base_quality_value_distribution_by_read_position_2.txt rawQuality.png cleanQuality.png 0 0
SOAPnuke is released under GPLv3. The latest source code is freely available at github.
- Chen Y, Chen Y, Shi C, et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience. 2018;7(1):1-6. doi:10.1093/gigascience/gix120 [PMID: 29220494]