Data structures and functions to draw biological macromolecules.
This addon is based on ESBTL, a Protein Data Bank (PDB) parser and data structure for the structural and geometric analysis of biological macromolecules.
ofApp.h
#pragma once
#include "ofMain.h"
#include "ofxMol.h"
class ofApp : public ofBaseApp
{
public:
void setup();
void draw();
void update();
protected:
OfxMol::System system;
ofMesh mesh;
};ofApp.cpp
#include "ofApp.h"
//--------------------------------------------------------------
void ofApp::setup()
{
ofBackground(0);
// setup system
system.setup(ofFile("ice.pdb").path());
mesh = system.getModel(0).atomsMesh(0.5f);
}
//--------------------------------------------------------------
void ofApp::update()
{
ofSetWindowTitle(ofToString(ofGetFrameRate(), 0));
}
//--------------------------------------------------------------
void ofApp::draw()
{
ofEnableDepthTest();
ofPushMatrix();
ofTranslate(ofGetWidth()/2.0, ofGetHeight()/2.0);
ofScale(40.0, 40.0);
ofRotate(ofGetElapsedTimef()*1.5,0,1,1);
// draw atoms
ofSetColor(255);
mesh.draw();
ofPopMatrix();
ofDisableDepthTest();
// log info
ofSetColor(255);
ofDrawBitmapString(system.getModel(0).log(),10,20);
}
void ofApp::update()
{
for (int i = 0; i < model.number_of_atoms(); i++)
{
OfxMol::Atom atom = model.getAtom(i);
}
}Copyright (C) 2015 Davide Rambaldi
This program is free software: you can redistribute it and/or modify
it under the terms of the GNU General Public License as published by
the Free Software Foundation, either version 3 of the License, or
(at your option) any later version.
This program is distributed in the hope that it will be useful,
but WITHOUT ANY WARRANTY; without even the implied warranty of
MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the
GNU General Public License for more details.
You should have received a copy of the GNU General Public License
along with this program. If not, see http://www.gnu.org/licenses/.
-
Install and link boost
This addon DO NOT include boost, because probably you have it installed in your system somewhere.
My suggestion to install boost on Mac is homebrew:
brew install boostAfter installation you should link the includes directory where boost is installed:
For homebrew the directory is
/usr/local/include.In XCode:
Build Settings --> Header Search Path --> add:
/usr/local/include. -
Install addon
Clone the addon repository in your
openFrameworks/addons/folder. -
Run Examples
Open example-simple and change, if necessary, the boost header search path (see point 1).
Open Frameworks Version > 0.8.4
Chain and Residues not implemented yet
Basic version with mesh generation and some examples
A system provides access to the hierarchical data structure.
It may contain one or several models.
Each model is made of chains.
Chains are then made of residues, and residues are made of atoms.
There is a single system (OfxMol::System) that stores models (OfxMol::Models) and water_models.
The system is created using the function OfxMol::System::setup(ofFile file, SetupMode mode).
After setup the OfxMol::System contains a std::vector of OfxMol::Model named models and a std::vector of OfxMol::Model named water_models.
Possible SetupMode are:
| Mode | Atoms | Occupancy | Notes |
|---|---|---|---|
| SIMPLE | All atoms in one system | Accept all occupancy policies | Use this mode for simple molecules |
| ADVANCED | 2 systems the first one contains all heavy atoms that do not belong to a water molecule. the second one contains all containing heavy atoms of water molecules. Hydrogen atoms are discarded. Use water_models_begin and water_models_end to iterate over water models in second system |
Accept all occupancy policies | Use this mode for proteins and other big molecules |
| CHAIN | TODO | TODO | TODO |
Each OfxMol::Model contains:
- a vector of
OfxMol::Atoms - a vector of
OfxMol::Coarse_Atoms
In molecular dynamics, coarse graining consists of replacing an atomistic description of a biological molecule with a lower-resolution coarse-grained model that averages or smooths away fine details.
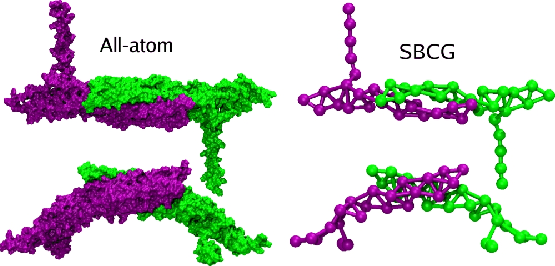
BAR domain homodimer. The two monomers are shown in green and purple. The all-atom structure is shown on the left, and an example of a coarse-graining structure is shown on the right. Both all-atom and SBCG structures are shown from the top and from the side.
See also: Coarse-graining in wikipedia
Install pymol to open and resave the molecule with Save Molecule in a new PDB files. This usually fix the PDB problems.