Home
Note, as of March 2024, TransDecoder is not being actively supported by its developer. Please continue to use as it fits your needs.
TransDecoder identifies candidate coding regions within transcript sequences, such as those generated by de novo RNA-Seq transcript assembly using Trinity, or constructed based on RNA-Seq alignments to the genome using StringTie.
TransDecoder is applied to an entire transcriptome for a single organism involving thousands of transcript sequences as input.
TransDecoder is unlikely to work if you provide a small number of sequences as input, as it requires training a species-specific model based on hundreds of candidates derived from the inputs.
TransDecoder identifies likely coding sequences based on the following criteria:
-
a minimum length open reading frame (ORF) is found in a transcript sequence
-
a log-likelihood score similar to what is computed by the GeneID software is > 0.
-
the above coding score is greatest when the ORF is scored in the 1st reading frame as compared to scores in the other 2 forward reading frames.
-
if a candidate ORF is found fully encapsulated by the coordinates of another candidate ORF, the longer one is reported. However, a single transcript can report multiple ORFs (allowing for operons, chimeras, etc).
-
a PSSM is built/trained/used to refine the start codon prediction.
-
optional the putative peptide has a match to a Pfam domain above the noise cutoff score.
The latest release of TransDecoder can be found here.
No compilation required - written in Perl, the favored scripting language of bioinformaticians back in the day.
The following Perl modules are needed and can be installed using cpanm like so:
curl -L https://cpanmin.us | perl - App::cpanminus
cpanm install DB_File
cpanm install URI::Escape
TransDecoder is also available for running via Docker or Singularity and includes BLAST+ and HMMER executables.
Note, TransDecoder is now available on Galaxy

The 'TransDecoder' utility is run on a fasta file containing the target transcript sequences. The simplest usage is as follows:
TransDecoder.LongOrfs -t target_transcripts.fasta
By default, TransDecoder.LongOrfs will identify ORFs that are at least 100 amino acids long. You can lower this via the '-m' parameter, but know that the rate of false positive ORF predictions increases drastically with shorter minimum length criteria.
If the transcripts are oriented according to the sense strand, then include the -S flag to examine only the top strand. Full usage info is below.
Include --complete_orfs_only if you want to exclude partials, but note that the chosen start codon may not be correct given that in the case of a 5' partial, the ORF extends further upstream. Also, 3' partials and full read-thru (no start and no stop) transcript sequences will be excluded.
Optionally, identify ORFs with homology to known proteins via blast or pfam searches.
See Including homology searches as ORF retention criteria section below.
TransDecoder.Predict -t target_transcripts.fasta [ homology options ]
The final set of candidate coding regions can be found as files '.transdecoder.' where extensions include .pep, .cds, .gff3, and .bed
The process here is identical to the above with the exception that we must first generate a fasta file corresponding to the transcript sequences, and in the end, we recompute a genome annotation file in GFF3 format that describes the predicted coding regions in the context of the genome.
Construct the transcript fasta file using the genome and the transcripts.gtf file like so:
util/gtf_genome_to_cdna_fasta.pl transcripts.gtf test.genome.fasta > transcripts.fasta
Next, convert the transcript structure GTF file to an alignment-GFF3 formatted file (this is done only because our processes operate on gff3 rather than the starting gtf file - nothing of great consequence). Convert gtf to alignment-gff3 like so, using cufflinks GTF output as an example:
util/gtf_to_alignment_gff3.pl transcripts.gtf > transcripts.gff3
Now, run the process described above to generate your best candidate ORF predictions:
TransDecoder.LongOrfs -t transcripts.fasta
(optionally, identify peptides with homology to known proteins)
TransDecoder.Predict -t transcripts.fasta [ homology options ]
And finally, generate a genome-based coding region annotation file:
util/cdna_alignment_orf_to_genome_orf.pl \
transcripts.fasta.transdecoder.gff3 \
transcripts.gff3 \
transcripts.fasta > transcripts.fasta.transdecoder.genome.gff3
The sample_data/ directory includes a 'runMe.sh' script that you can execute to demonstrate the entire process, starting from a cufflinks GTF file. Note, the typical use-case for TransDecoder is starting from a fasta file containing target 'Transcripts', however, in the case of genome analysis, transcripts are often inferred from annotation coordinates, such as in this Cufflinks GTF formatted file. In this example, transcript sequences are reconstructed based on the GTF annotation coordinates, and then TransDecoder is executed on that fasta file. We include an additional utility for converting the transcript ORF coordinates into genome-coordinates so these regions can be examined in the genomic context.
A working directory (ex. transcripts.transdecoder_dir/) is created to run and store intermediate parts of the pipeline, and contains:
longest_orfs.pep : all ORFs meeting the minimum length criteria, regardless of coding potential.
longest_orfs.gff3 : positions of all ORFs as found in the target transcripts
longest_orfs.cds : the nucleotide coding sequence for all detected ORFs
longest_orfs.cds.top_500_longest : the top 500 longest ORFs, used for training a Markov model for coding sequences.
hexamer.scores : log likelihood score for each k-mer (coding/random)
longest_orfs.cds.scores : the log likelihood sum scores for each ORF across each of the 6 reading frames
longest_orfs.cds.scores.selected : the accessions of the ORFs that were selected based on the scoring criteria (described at top)
longest_orfs.cds.best_candidates.gff3 : the positions of the selected ORFs in transcripts
Then, the final outputs are reported in your current working directory:
transcripts.fasta.transdecoder.pep : peptide sequences for the final candidate ORFs; all shorter candidates within longer ORFs were removed.
transcripts.fasta.transdecoder.cds : nucleotide sequences for coding regions of the final candidate ORFs
transcripts.fasta.transdecoder.gff3 : positions within the target transcripts of the final selected ORFs
transcripts.fasta.transdecoder.bed : bed-formatted file describing ORF positions, best for viewing using GenomeView or IGV.
To further maximize sensitivity for capturing ORFs that may have functional significance, regardless of coding likelihood score as mentioned above, you can scan all ORFs for homology to known proteins and retain all such ORFs. This can be done in two popular ways: a BLAST search against a database of known proteins, and searching PFAM to identify common protein domains. In the context of TransDecoder, this is done as follows:
After running TransDecoder.LongOrfs, you'll find a multi-fasta protein file called '${transcripts_file}.transdecoder_dir/longest_orfs.pep'. Search these candidate peptides for homology using approaches below:
Search a protein database such as Swissprot (fast) or Uniref90 (slow but more comprehensive) using BLAST+
An example command would be like so:
blastp -query transdecoder_dir/longest_orfs.pep \
-db uniprot_sprot.fasta -max_target_seqs 1 \
-outfmt 6 -evalue 1e-5 -num_threads 10 > blastp.outfmt6
If you have access to a computing grid, consider using HPC GridRunner for more efficient parallel computing.
Some prefer to use diamondblast instead of NCBI blast for greatly increased speed - but beware of potential differences in sensitivity.
Search the peptides for protein domains using Pfam. This requires hmmer3 and Pfam databases to be installed.
hmmsearch --cpu 8 -E 1e-10 --domtblout pfam.domtblout /path/to/Pfam-A.hmm transdecoder_dir/longest_orfs.pep
Both hmmscan and hmmsearch should work here, but hmmsearch is recommended due to much faster execution.
Just as with the blast search, if you have access to a computing grid, consider using HPC GridRunner.
The outputs generated above can be leveraged by TransDecoder to ensure that those peptides with blast hits or domain hits are retained in the set of reported likely coding regions. Run TransDecoder.Predict like so:
TransDecoder.Predict -t target_transcripts.fasta --retain_pfam_hits pfam.domtblout --retain_blastp_hits blastp.outfmt6
The final coding region predictions will now include both those regions that have sequence characteristics consistent with coding regions in addition to those that have demonstrated blast homology or pfam domain content.
GenomeView or IGV are recommended for viewing the candidate ORFs in the context of the genome or the transcriptome. Examples below show GenomeView in this context.
java -jar $GENOMEVIEW/genomeview.jar transcripts.fasta transcripts.fasta.transdecoder.bed
If you lack a genome sequence and are working exclusively with the target transcripts, you can load the transcript fasta file and the ORF predictions (bed file) into GenomeView (see below).
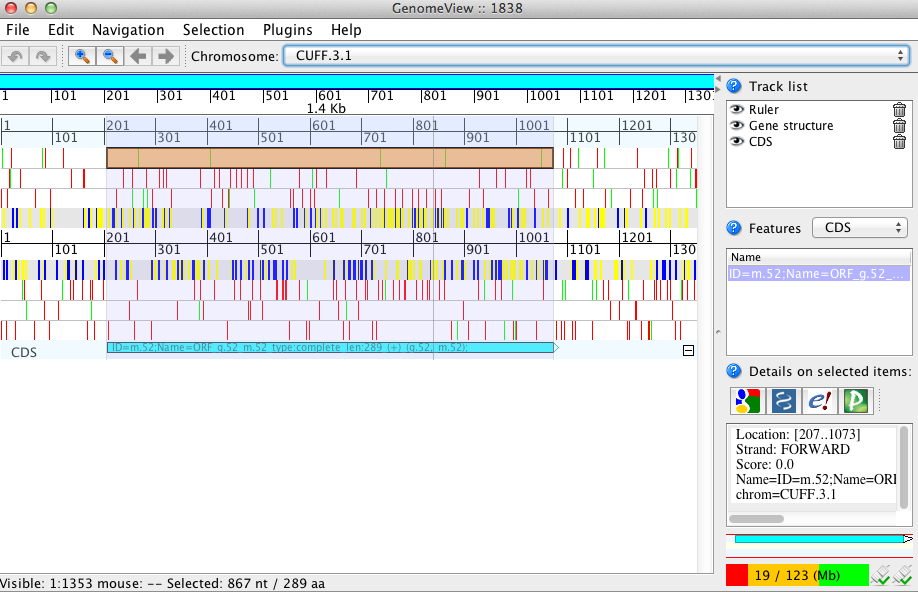
java -jar $GENOMEVIEW/genomeview.jar test.genome.fasta transcripts.bed transcripts.fasta.transdecoder.genome.bed
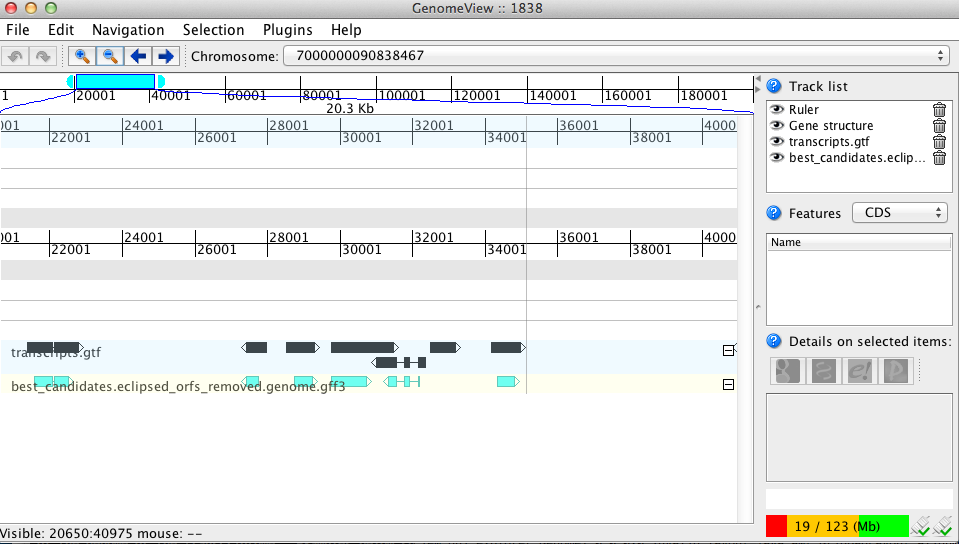
The original cufflinks-based transcript structures are shown in black, and the predicted coding regions are shown in cyan.
Join our TransDecoder google group at https://groups.google.com/forum/#!forum/transdecoder-users
There's no manuscript to cite for TransDecoder. Instead, simply cite the website as: 'Haas, BJ. https://github.com/TransDecoder/TransDecoder' and whichever software version you use.