Visualization of RNA secondary structure using a template visualization (currently Comparative RNA Web (CRW) Site postscripts and VARNA SVG formats are supported). The first version of Traveler was developed by Richard Elias (and still the absolulte majority of the code is his work) and the original repository should be accessible at https://github.com/rikiel/traveler .
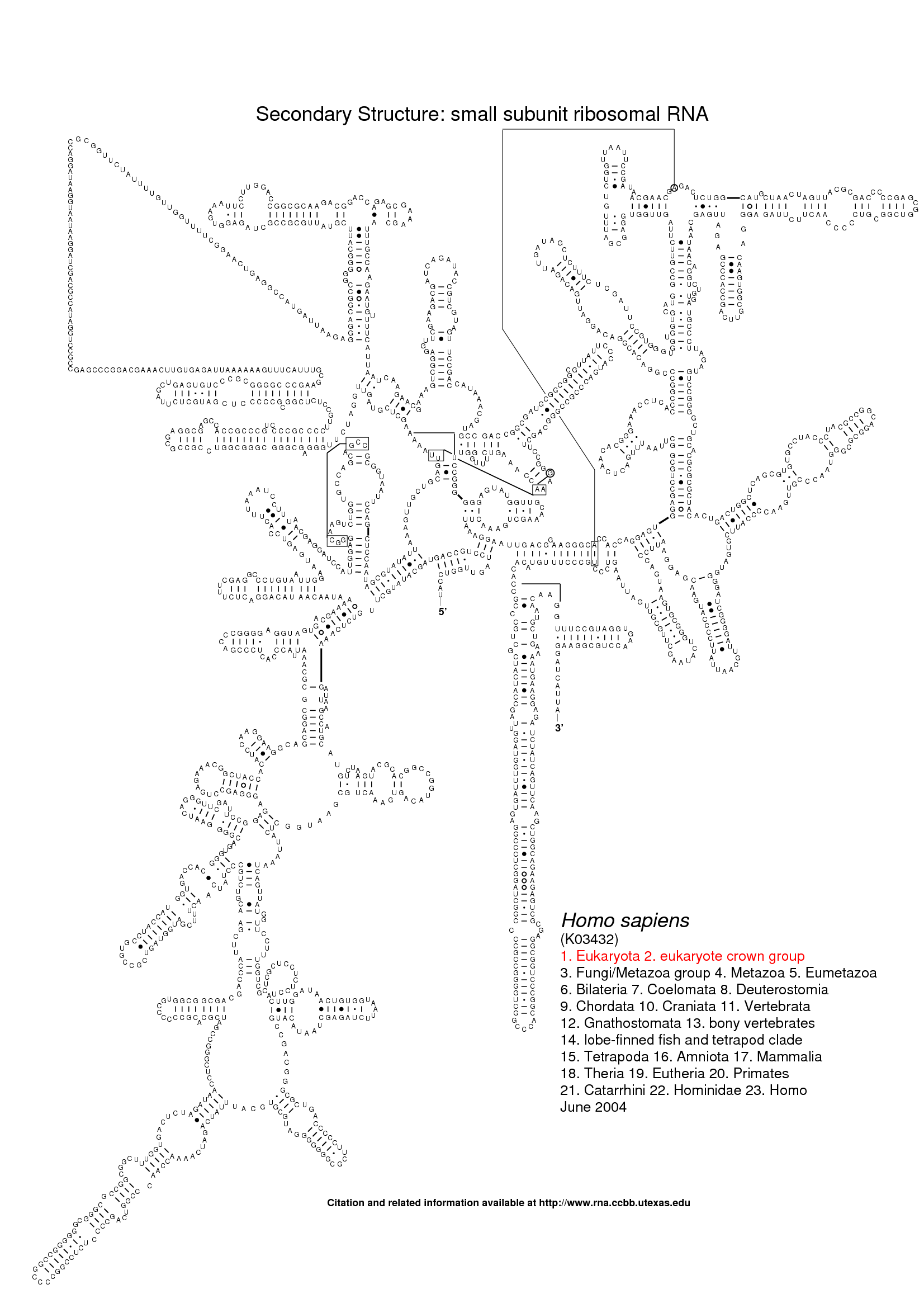
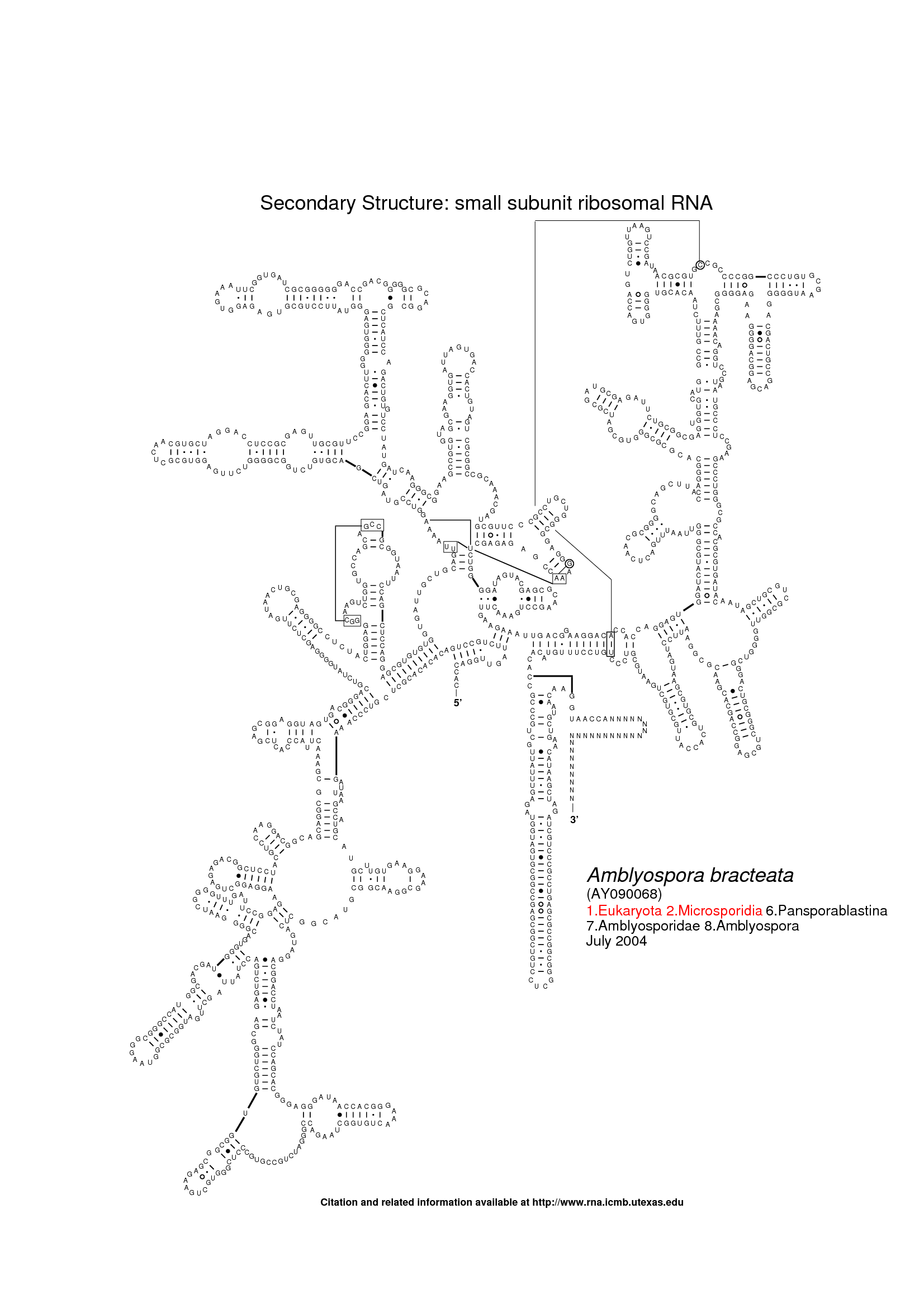
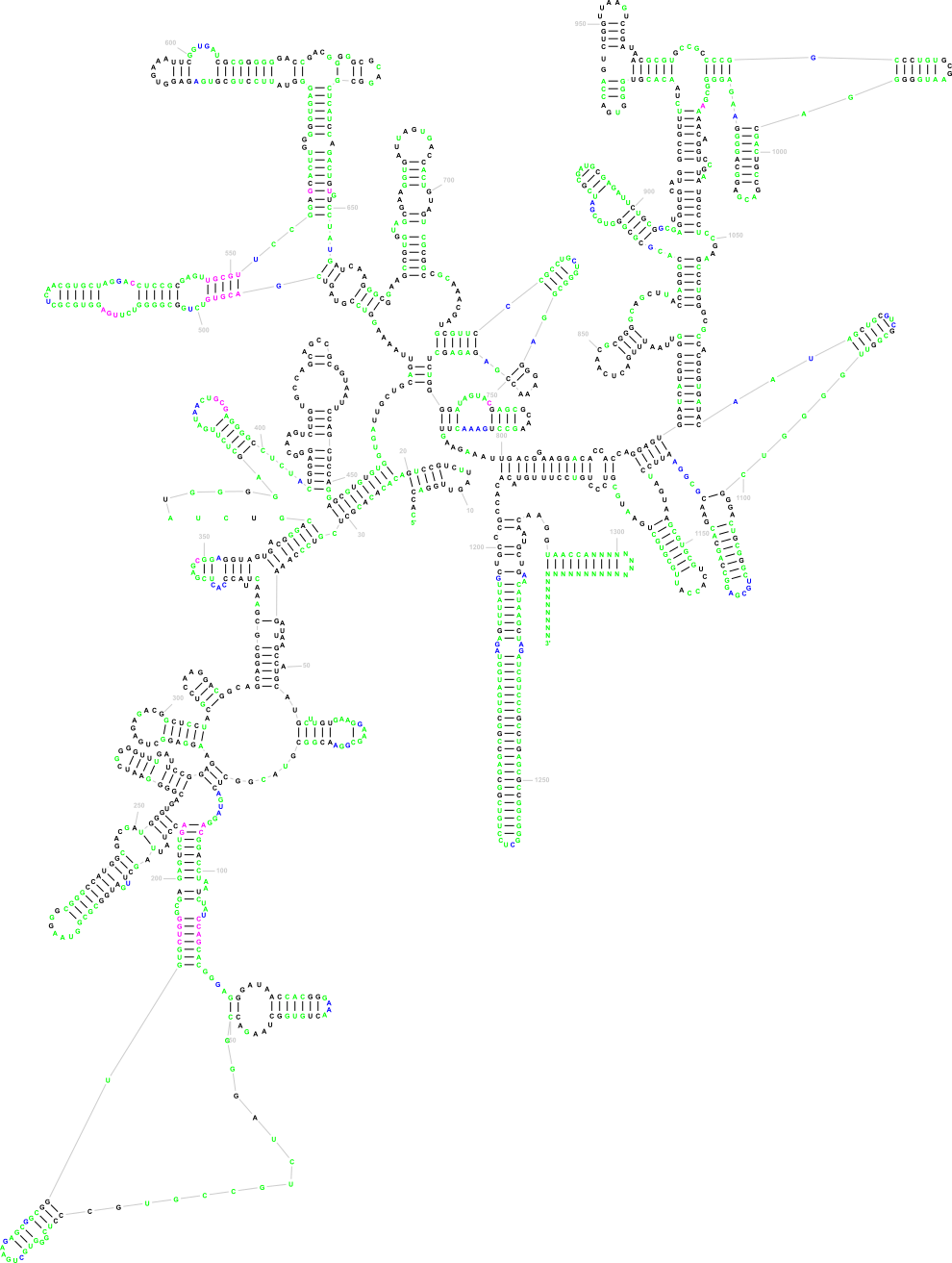
The following three images show the purpose of Traveler. The first image is the visualization of 18S human rRNA from CRW, follows the correct visualization of Amblyospora bracteata's (AB) 18s rRNA (as downloaded from CRW) and the third image shows how Traveler visualizes the AB's 18s when given its secondary structure together with human rRNA secondary structure and visualization in postscript as a template.
Traveler is used by RNACentral for visualization of all the secondary structures available in RNACentral. See the R2DT project for details.
$ mkdir test
$ bin/traveler \
--target-structure data/fungi/d.16.e.A.bracteata.fasta \
--template-structure data/metazoa/human.ps data/metazoa/human.fasta \
--all test/ab_from_human
- gcc with support of c++11
- Python3 for running utils (e.g. conversion from RNA2D-data-schema JSON output) to SVG
Use git clone https://github.com/cusbg/traveler to download project
cd traveler/src
make build
cd ..
The binaries will be copied into traveler/bin. To navigate there from the src directory use: cd ../bin
-
Download the source code and
cdinto the traveler directory. -
Build an image:
docker build . -t traveler
- Run the container:
docker run -v "$PWD":/data -it traveler
The traveler executable is available in the PATH, and the current directory is mounted in the container under /data.
traveler [-h|--help]
traveler [OPTIONS] <STRUCTURES>
STRUCTURES:
<-gs|--target-structure> DBN_FILE
<-ts|--template-structure [--file-format FILE_FORMAT]> IMAGE_FILE DBN_FILE
DBN_FILE (Varna/DotBracketNotation) is in format like in example below
IMAGE_FILE* - visualization of template molecule, type of file can be specified by FILE_FORMAT argument
OPTIONS:
[-a|--all] [--overlaps] OUT_PREFIX
# computes mapping (TED) and outputs target layout as an .svg image and .xml file to files with prefix OUT_PREFIX
# with the optional --overlaps argument, overlaps in the layout are identified and highlited
[-t|--ted <FILE_MAPPING_OUT>]
# runs mapping (TED) only and saves mapping table to FILE_MAPPING_OUT file
[-d|--draw] [--overlaps] FILE_MAPPING_IN OUT_PREFIX
# use mapping in FILE_MAPPING_IN and outputs layout as an .svg image and .xml file to files with prefix OUT_PREFIX
# if optional argument --overlaps is present overlaps in the layout are identified and highlighted
[-r|--rotate] If switched on, Traveler tries to rotate hairpins to minimize the number of overlaps. In some
cases, this can lead to a more convoluted layout and therefore this features is turned off by default.
[-n|--numbering] NUMBERING_DEFINITION
# Allows to specify residues which will have number information next to it in the resulting diagram.
# The format allows to specify list of residue indexes and interval so that every residue index which
# is modulo interval == 0 will be labeled. The default value is "10,20,30-50", i.e. residues with indexes
# 10, 20, 30 and every 50th residue will be labeled.
[-l|--labels-template] Uses template labels for numbering. This
is useful, for example, in case of tRNA where users are used to the Sprinzl positions. Here, for instance, the 21st residue of a particular tRNA
is Sprinzl position 20a. So if the 21st residue is mapped onto a target residue with visible nubmer (e.g. 20 by default),
that label should show 20a irrespective of its position in the target.
[-v|--verbose] Prints information about the computation and othere details (such as number of overlaps,
when overlap switch is turned on)
COLOR CODING:
Traveler uses the following color coding of nucleotides:
* Magenta - inserted bases
* Green - edited bases - e.g. the template has an adenosine at a position while the target has a cytosine at the same position and therefore cytosine will be colored green in the resulting layout)
* Blue - reinserted bases - happens when traveler needs to redraw simple structures like hairpins (for example due to the change in the number of bases)
* Brown - rotated parts - similar situation to reinserted bases, but takes place when redrawing a multibranch loop (in that case all branches need to be rotated to lie on a circle)
Three types of template IMAGE_FILE are currectly supported by Traveler:
- crw: PostScript from CRW (default)
- varna: VARNA format of SVG images produced by tool VARNA
- traveler: Traveler intermediate format
Traveler's intermediate format is a simple XML which contains an ordered list of nucleotides (information about lines is not necessary since lines connecting backbone are defined by the order of nucleotides and base pairing lines are defined by the secondary structure).
<structure>
<point x="138.3" y="-367" b="U" />
<point x="138.3" y="-359" b="A" />
.
.
.
<point x="278.1 y="-504.6" b="A" />
</structure>
Additionally, the format can include position labels which can then be used in the target numbering (see the -l argument).
<structure>
<point x="69.55" y="39.53" b="G" numbering-label="1"/>
<point x="69.91" y="46.89" b="N" numbering-label="2"/>
<point x="69.97" y="54.66" b="Y" numbering-label="3"/>
<point x="69.76" y="62.22" b="C" numbering-label="4"/>
<point x="69.91" y="69.57" b="N" numbering-label="5"/>
<point x="69.91" y="77.13" b="N" numbering-label="6"/>
<point x="69.76" y="84.89" b="R" numbering-label="7"/>
<point x="62.94" y="88.16" b="U" numbering-label="8"/>
<point x="57.38" y="93.50" b="G" numbering-label="9"/>
<point x="54.12" y="100.32" b="G" numbering-label="10"/>
<point x="46.77" y="100.32" b="C" numbering-label="11"/>
<point x="39.36" y="100.12" b="N" numbering-label="12"/>
<point x="31.86" y="100.32" b="Y" numbering-label="13"/>
<point x="26.52" y="94.97" b="A" numbering-label="14"/>
<point x="19.10" y="93.49" b="A" numbering-label="15"/>
<point x="11.91" y="96.38" b="U" numbering-label="16"/>
<point x="7.50" y="102.66" b="G" numbering-label="18"/>
<point x="7.50" y="110.22" b="G" numbering-label="19"/>
<point x="12.06" y="116.30" b="N" numbering-label="20"/>
<point x="19.05" y="119.19" b="N" numbering-label="20a"/>
<point x="26.52" y="117.92" b="A" numbering-label="21"/>
...
<structure>
Other RNA structure extractors can be implemented and specified by the FILE_FORMAT argument.
Traveler accepts FASTA-like file format (see Example 0) for the description of the template structucture. You can prepare it manually, or, if you are using CRW as the source of templates, you can download the pseudoknot-free version version of the structure in the bpseq format and use it as the input to the bpseq2fasta Python script which can be found in the Utilities directory.
The file needs to contain three lines: moelcule description line (starts with the > symbol), sequence line, structure line. Should you have a sequence in a FASTA file with sequence spanning multiple lines, you can use the following script to obtain single-line sequence:
awk 'BEGIN {ORS=""}!/^>/{print}' sequence.fasta
$ cat data/metazoa/mouse.fasta
>mouse
UACCUGGUUGAUCCUGCCAGUAGCAUAUGCUUGUCUCAAAGAUUAAGCCAUGCAUGUCUAAGUACGCACGGCCGGUACAG
UGAAACUGCGAAUGGCUCAUUAAAUCAGUUAUGGUUCCUUUGGUCGCUCGCUCCUCUCCUACUUGGAUAACUGUGGUAAU
. . .
...(((((.......))))).((((((((((.(((((((((.....(((.(((..((...(((....((..........)
)...)))))......(((......((((..((..((....(((..................((((....(((((((....
. . .
FASTA (Varna/DBN) file should contain line starting with '>' and name of molecule, without any blank character
other lines are filled with LABELS and BRACKETS in dot-bracket notation of secondary structure pairing
match-tree must contain both LABELS and BRACKETS, templated-tree need only BRACKETS
Example 1a: Visualize mouse 18S rRNA using human 18S rRNA as template using CRW ps image as the template layout
$ mkdir test
$ bin/traveler \
--target-structure data/metazoa/mouse.fasta \
--template-structure data/metazoa/human.ps data/metazoa/human.fasta \
--all test/mouse_from_human
Outputs a SVG and JSON file (see Example3 below for details)
Example 1b: Visualize mouse 18S rRNA using human 18S rRNA as template using Traveler's XML input format as the template layout.
$ mkdir test
$ bin/traveler \
--target-structure data/metazoa/mouse.fasta \
--template-structure --file-format traveler data/metazoa/human.xml data/metazoa/human.fasta \
--all test/mouse_from_human_xml_input
Example 2: Compute TED distance and mapping between human 18S rRNA (template) and mouse 18S rRNA (target).
$ mkdir test
$ bin/traveler \
--target-structure data/metazoa/mouse.fasta \
--template-structure data/metazoa/human.ps data/metazoa/human.fasta \
--ted test/mouse_from_human.map
$ mkdir test
$ bin/traveler \
--target-structure data/metazoa/mouse.fasta \
--template-structure data/metazoa/human.ps data/metazoa/human.fasta \
--draw --overlaps test/mouse_from_human.map test/mouse_from_human
# checks also if output molecule has overlaps and draws them in output image, but this information is not available in the json file (see below)
The above example enerates 2 files - 1 .svg file (see COLOR CODING section) and 1 .json file in the
RNA2D data schema format. The json file can then be
converted into a SVG format using the json2svg utility (see the utils directory for more arguments):
python3 utils/json2svg.py -i test/mouse_from_human.json -o test/mouse_from_human.json.svgOptions --ted and --draw serve for separatation of mapping and visualization since TED computation and on the other hand, Traveler allows for multiple output visualization (coloring, overlaps).
Traveler supporst two types of input images - crw and varna. There are three steps that need to done when one wants to support other image types.
- You need to implement
extractorinterface and its methodextract. The method accepts nucleotides (the primary structure) and their position in the image (points) from given file. - In the class, you need to implement method
get_type, that should only return extractor's type that is used in IMAGE_FILE argument. - Add your new extractor to method
extractor.get_all_extractors()
For more ideas, how it should be implemented, see usage of crw_extractor and varna_extractor.
As shown in the examples above, the Traveler procedure can be split into two phases - structure mapping phase and layout phase. In the mapping phase, both target and template RNA structures are turned into tree representations and tree edit distance is employed to find a mapping between the nodes of the two trees. The description of how the base-pairing information can be turned to a tree representation is described in the Traveler paper. Please note, that the numbering of the nodes in the tree is post order. Also, the numbering starts from 0, but the first node is dedicated to the 5'3' "pair". So numbering of the "real" residues (either unpaired or base-paired nucleotides) starts with 1. The output of the mapping phase is a list of pairs indicating which nodes are mapped onto each other and which need to be inserted/deleted. Follows an example of such mapping. Please note that insertions/deletions correspond to lines containing 0 either in the first or second position. The first column corresponds to the template and the second to the target structure:
DISTANCE: 4
0 12
0 13
1 1
2 2
3 3
4 4
5 5
6 6
7 7
8 8
9 9
10 10
11 11
12 14
13 15
14 16
15 0
16 17
17 18
18 19
19 20
20 21
21 22
22 23
23 24
24 25
25 26
26 27
27 28
28 29
29 30
30 31
31 32
32 33
33 34
34 35
35 36
36 37
37 0
38 38
39 39
40 40
41 41
42 42
43 43
44 44
45 45
46 46
47 47
48 48
49 49
50 50
51 51
52 52
53 53
54 54
55 55
56 56
57 57
58 58
59 59
60 60
The tree mapping procedure implemented in Traveler might not always lead to optimal mapping. That is especially true when you are predicting the secondary structure of the target via homology modeling and you thus know the correct mapping between the target and template. In such case, it might be advantageous to provide the mapping manually. That is exactly the case of using Traveler in the R2DT pipeline used by RNAcentral where the structure is folded with Infernal. In such case one can use the secondary structure produced by Infernal and stored in the Stockholm file format to generate the corresponding sequence and, subsequently, tree mapping. The following example makes use of the scripts from the Infernal scripts repository:
perl ~/git/jiffy-infernal-hmmer-scripts/ali-pfam-lowercase-rf-gap-columns.pl target.stk > target.indi.pfam.lc.stk
perl ~/git/jiffy-infernal-hmmer-scripts/ali-pfam-sindi2dot-bracket.pl -l -n -w -a -c target.indi.pfam.lc.stk > target.traveler.afa
python3 ~/git/traveler/utils/infernal2mapping.py -i target.traveler.afa > target.map
~/git/traveler/bin/traveler --target-structure target.fasta --template-structure --file-format traveler template.layout template.fasta --draw target.map target
In case of homology-based modeling, RNA structure is predicted in two steps; i) carrying over structure within conserved regions from template, and ii) de-novo prediction of the unconserved regions. Typically, one have substantially more confidence in the predictions in the conserved regions. Therefore, it make sense to be able to visualy distinguish base pairs predicted de novo from those which were copied over from the template. This information can be provided in the template FASTA file in the optional fourth line.
>structure predicted via constraint folding
CAUCCGCAGGUGCCCCUAGAAAAAAAUUGUGCCUAGGACCCCCCUGCGCGAGGGGUAG
((.(((.(((....))).(((.....))...((..((.....))..)))...))).))
---*--------------------------------------------------*---
In the above structure, the CG basepair was predicted de novo and not carried over from the template. This will be depicted in the resulting SVG as a dotted gray line between the respective bases.

Traveler supports visualziation of pseduoknots , but does not consider them during the matching process. Specifically, when parsing the target structure, pseudoknot pairs are "depaired" and the matching is carried out as if the corresponding residues were unpaired. Then, after the layout is generated, the pseduknots are reintroduced into the final visualization.
Pseudoknots are visualized as straight gray lines with some level of opacity so that they do not clutter the layout. Still to decrease the clutter possibly caused by pseudoknots, continous regions of pseudoknots are grouped together. In such case, the pseudoknotted residues that follow in sequence gain gray background and only the residues of the first pseudoknot are connected.
To support pseduoknots of (efficiently) arbitrary depth, the following charcters can be used to mark pseudoknot
pairs in the dot-bracket notation: []{}AaBbCcDdEeFfGgHhIiJjKkLlMmNnOoPpQqRrSsTtUuVvWwXxYyZz. Odd characters
denote opening brackets which even characters denote closing bracket. Follows an example of a target with second-level
pseudoknots:v
>2 BPs added to stem
CAUCCGCAGGUGCCCCUAGAAAAAAAUUGUGCCUAGGACCCCCCUGCGCGAGGGGUAG
((.(((A(((a...))).(((AAABA))b..((..((aaa.a))..)))...))).))
---*--------------------------------------------------*---
To get the layout with the above target and the base template from the test directory( base.fasta , base.svg), run the following command:
{kind=link}
traveler --target-structure add_bp_to_stem.fasta --template-structure --file-format varna base.svg base.fasta --all out
You should end up with the following layout:

If you use Traveler in your research, please cite:
- Elias, R., & Hoksza, D. (2017). TRAVeLer: a tool for template-based RNA secondary structure visualization. BMC Bioinformatics, 18(1), 487.
![]()
Traveler is a part of services provided by ELIXIR – European research infrastructure for biological information. See services provided ELIXIR's Czech Republic Node.