
GET_PHYLOMARKERS (Vinuesa et al., 2018) is an open-source software package for selecting optimal markers for microbial phylogenomics and species tree estimation. It implements a bioinformatics pipeline to filter core-genome gene clusters computed by the companion package GET_HOMOLOGUES, and selects only those with optimal attributes for phylogenetic inference using maximum likelihood (ML). The multiple sequence alignments of the filtered loci are concatenated into a supermatrix to estimate a species tree using the state-of-the-art fast ML tree searching algorithms FastTree or IQ-TREE. It also estimates ML and parsimony trees from the pan-genome matrix, including unsupervised learning methods. We have also tested it successfully with plant coding sequences (details here).
{kind=link}
Starting with release 2.0.0 (2022-11-20), GET_PHYLOMARKERS also computes a concatenation-independent species tree from the ML gene trees estimated from top-scoring alignments using ASTRAL-III.
Release 2.1.0 (2024-03-31) introduced maximal matched-pairs tests to assess violations of the data to the Stationarity, Reversibility, and Homogeneity (SRH) assumptions made by maximum-likelihood phylogenetic models, as implemented in IQ-TREE. Additionally, ASTRAL-IV is used to estimate the species tree directly from the filtered ML source gene trees, which computes terminal and internal branch lengths in substitution-per-site units.
Release 2.2.0 (v2.2.0, 2024-04-14) introduced new features, most notably:
-
significant extension of run mode 2 (
run_get_phylomarkers_pipeline.sh -R 2) for population genetics (multiple sequences from the same species), computing a population tree from a SNP matrix of top-scoring, neutral alignments. -
Reporting of a detailed numeric overview of the different filtering steps
-
run_get_phylomarkers_pipeline.shnow also calls the C binary WEIGHTED-ASTRAL to estimate a species tree using as input the filtered gene trees estimated by iqtree2 or FastTree -
Complex protein mixture models are used for concatenated protein alignments
Release 2.2.1 (2024-04-16) includes a static binary of snp-sites, which is called by run_get_phylomarkers_pipeline.sh >= v2.8.1.0_2024-04-15 under run mode 2 (-R 2, population genetics) to generate SNP matrices in FASTA and VCF formats from the concatenated alignments of filtered, highly informative, and neutral loci. The FASTA SNP matrix is used for estimating a ML population tree. Thanks to Alfredo Hernández @ccg_unam for compiling snp-sites-static.
- This release was used to build the latest Docker GET_PHYLOMARKERS image (20240418) ready to pull from Docker Hub (
docker pull vinuesa/get_phylomarkers:latest. This is a significantly lighter (2G.0B) image than the previous one (v20240414 (2.09GB), because several unnecessary R packages were removed. On Dockerhub, you will find detailed instructions on installing and configuring the Docker client on your machine, pulling the latest image, and running the containerized instance of the GET_PHYLOMARKERS pipeline.
GET_PHYLOMARKERS has a detailed manual and step-by-step tutorials document the software and help the user to get up and running quickly. For convenience, html and markdown versions of the documentation material are available.
For detailed instructions and dependencies please check INSTALL.md.
A GET_PHYLOMARKERS Docker image is available, as well as an image bundling GET_PHYLOMARKERS + GET_HOMOLOGUES, ready to use. Detailed instructions for setting up the Docker environment are provided in INSTALL.md. How to run container instances with the test sequences distributed with GET_PHYLOMARKERS is described in the tutorial.
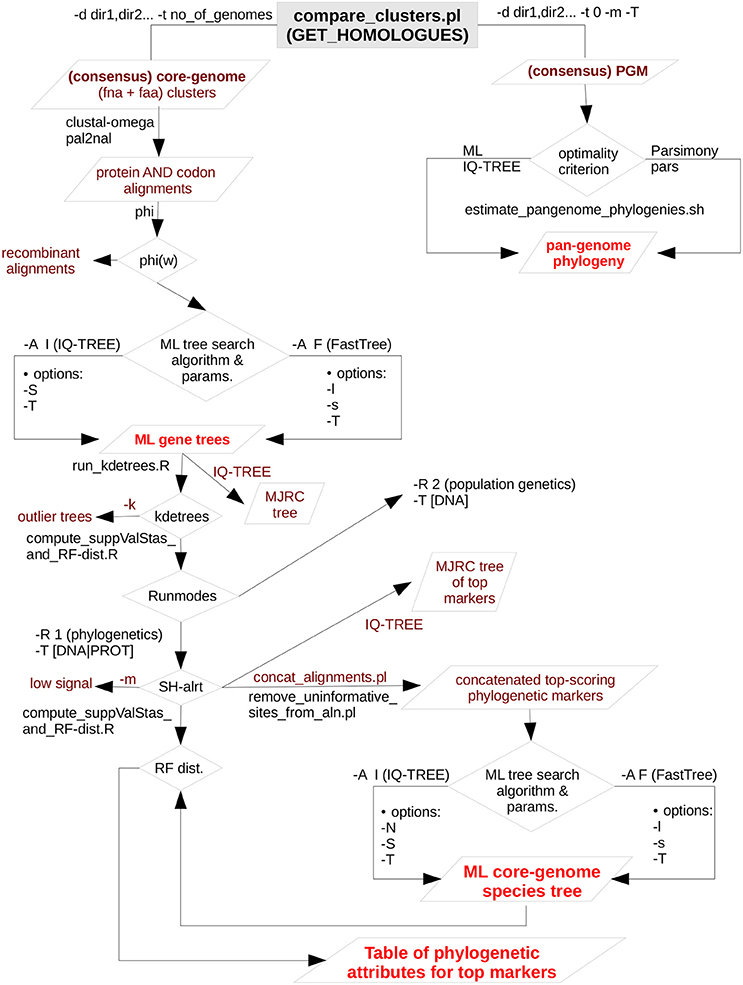
GET_PHYLOMARKERS (Vinuesa et al. 2018) implements a series of sequential filters (detailed below) to selects markers from the homologous gene clusters produced by GET_HOMOLOGUES with optimal attributes for phylogenomic inference. It estimates gene-trees and species-trees under the maximum likelihood (ML) optimality criterion using state-of-the-art fast ML tree searching algorithms. The species tree is estimated from the supermatrix of concatenated, top-scoring alignments that passed the quality filters outlined in the figures below and explained in detail in the manual and publication.


Figure 1A. Simplified flow-chart of the GET_PHYLOMARKERS pipeline showing only those parts used and described in this work. The left branch, starting at the top of the diagram, is fully under control of the master script run_get_phylomarkes_pipeline.sh. The names of the worker scripts called by the master program are indicated on the relevant points along the flow, as detailed in the manual. The image corresponds to Fig. 1 of Vinuesa et al. 2018.
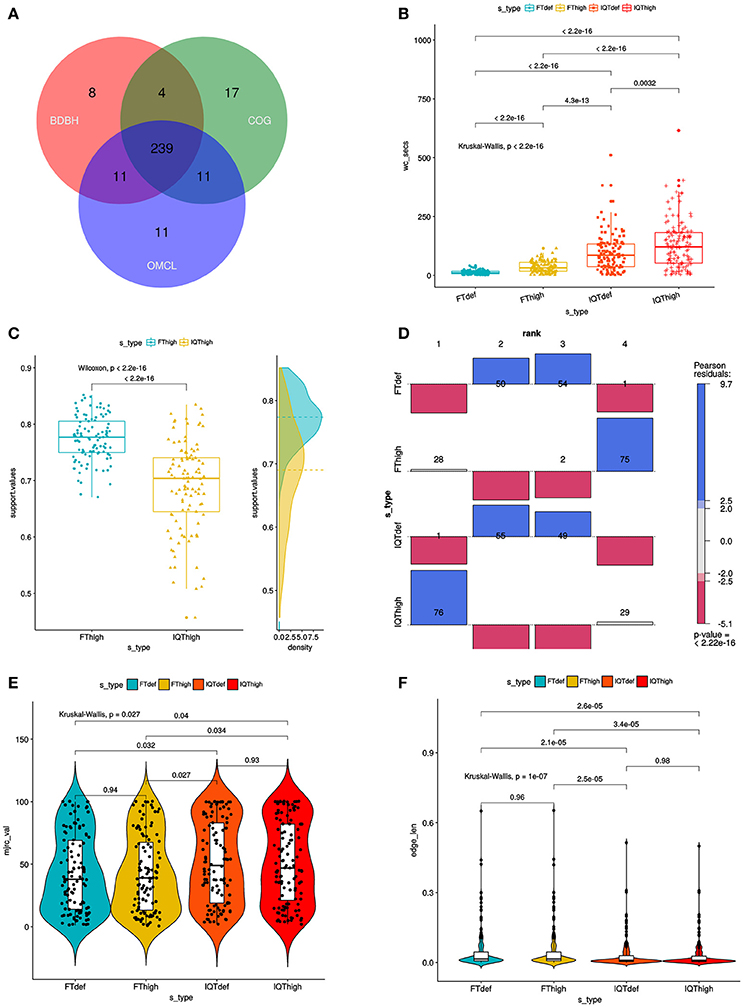
Figure 1B. Combined filtering actions performed by GET_HOMOLOGUES and GET_PHYLOMARKERS to select top-ranking phylogenetic markers to be concatenated for phylogenomic analyses, and benchmark results of the performance of the FastTree (FT) and IQ-TREE (IQT) maximum-likelihood (ML) phylogeny inference programs. The image corresponds to Fig. 3 of Vinuesa et al. 2018.
{kind=link}
GET_HOMOLOGUES is a genome-analysis software package for microbial pan-genomics and comparative genomics originally described in the following publications:
More recently we developed GET_HOMOLOGUES-EST, which can be used to cluster eukaryotic genes and transcripts, as described in Contreras-Moreira et al, Front. Plant Sci. 2017.
If GET_HOMOLOGUES_EST is fed both .fna and .faa files of CDS sequences it will produce identical output to that of GET_HOMOLOGUES and thus can be analyzed with GET_PHYLOMARKERS all the same.
GET_PHYLOMARKERS is primarily tailored towards selecting CDSs (gene markers) to infer DNA-level phylogenies of different species of the same genus or family. It can also select optimal markers for population genetics, when the source genomes belong to the same species (Vinuesa et al. 2018). For more divergent genome sequences, classified in different genera, families, orders or higher taxa, the pipeline should be run using protein instead of DNA sequences.


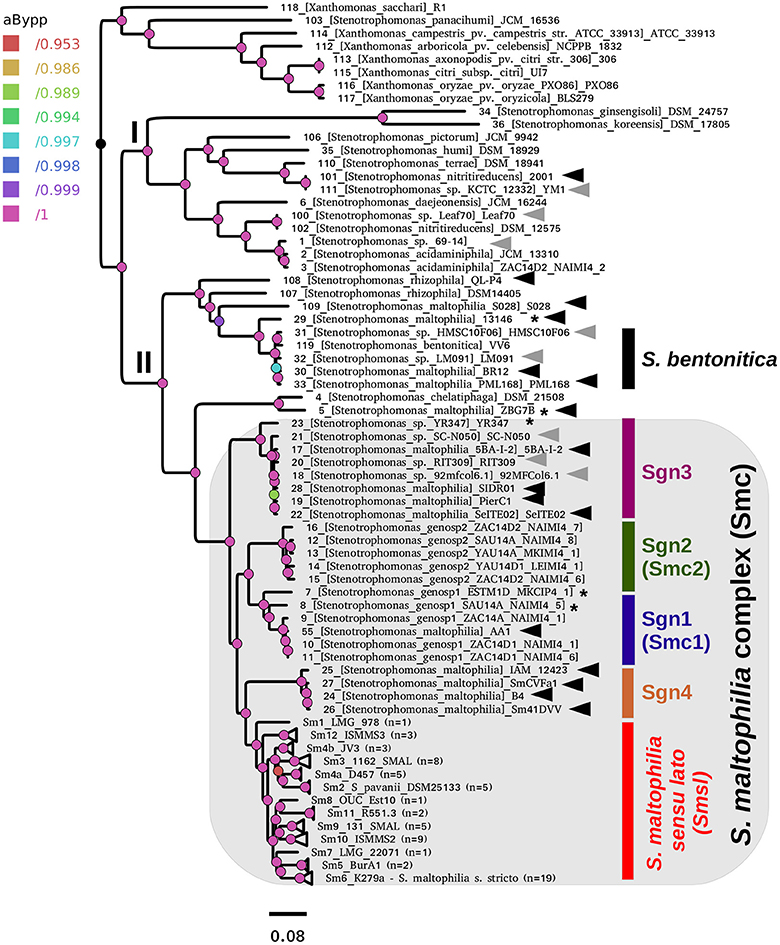
Figure 2A. Best maximum-likelihood core-genome phylogeny for the genus Stenotrophomonas found in the IQ-TREE search, based on the supermatrix obtained by concatenation of 55 top-ranking alignments. The image corresponds to Fig. 5 of Vinuesa et al. 2018.
{kind=link}
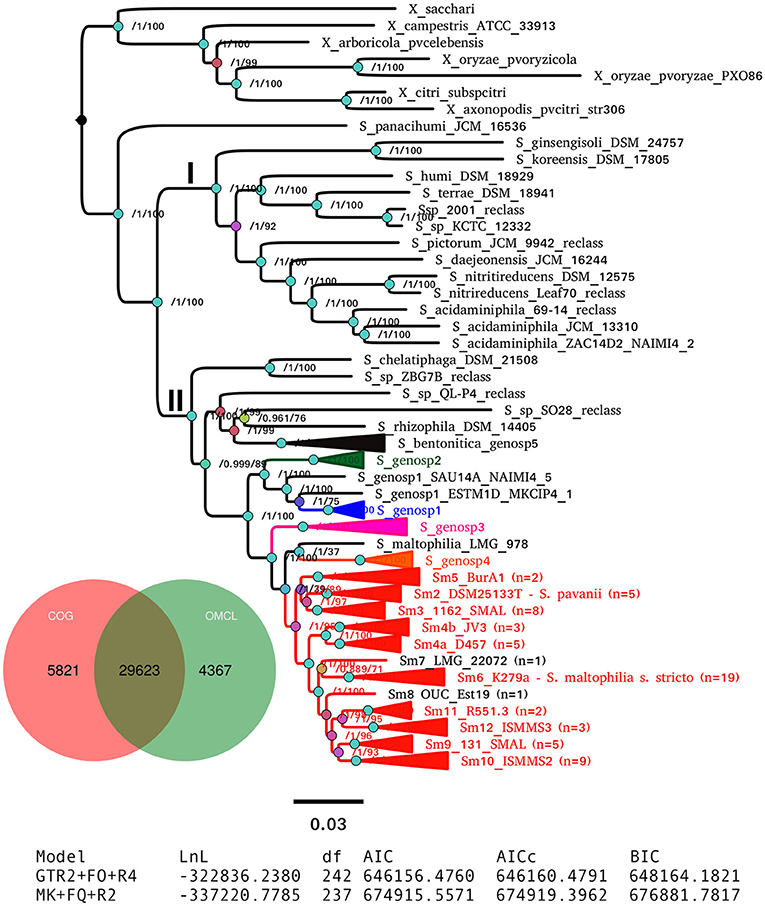
Figure 2B. Maximum-likelihood pan-genome phylogeny estimated with IQ-TREE from the consensus pan-genome clusters displayed in the Venn diagram. Clades of lineages belonging to the S. maltophilia complex are collapsed and are labeled as in Figure 2A. Numbers on the internal nodes represent the approximate Bayesian posterior probability/UFBoot2 bipartition support values (see methods). The tabular inset shows the results of fitting either the binary (GTR2) or morphological (MK) models implemented in IQ-TREE, indicating that the former has an overwhelmingly better fit. The scale bar represents the number of expected substitutions per site under the binary GTR2+F0+R4 substitution model. The image corresponds to Fig. 6 of Vinuesa et al. 2018.
{kind=link}
Please, follow the links for a detailed manual and tutorials, including a graphical flowchart of the pipeline and explanations of the implementation details. See also our plant tutorial.
Pablo Vinuesa, Luz-Edith Ochoa-Sanchez and Bruno Contreras-Moreira (2018). GET_PHYLOMARKERS, a software package to select optimal orthologous clusters for phylogenomics and inferring pan-genome phylogenies, used for a critical geno-taxonomic revision of the genus Stenotrophomonas. Front. Microbiol. | doi: 10.3389/fmicb.2018.00771
Published in the Research Topic on "Microbial Taxonomy, Phylogeny and Biodiversity" http://journal.frontiersin.org/researchtopic/5493/microbial-taxonomy-phylogeny-and-biodiversity
- Source sode is freely available from GitHub and released under a GPLv3-like license.
- Docker images ready to pull
The code is developed and maintained by Pablo Vinuesa at CCG-UNAM, Mexico and Bruno Contreras-Moreira at EEAD-CSIC, Spain.
We thank Alfredo J. Hernández and Víctor del Moral at CCG-UNAM for technical support with server administration.
We gratefully acknowledge the funding provided over the years by DGAPA-PAPIIT/UNAM (grants IN201806-2, IN211814, IN206318, and IN216424) and CONAHCyT-Mexico (grants P1-60071, 179133 and A1-S-11242) to Pablo Vinuesa, as well as the Fundación ARAID,Consejo Superior de Investigaciones Científicas (grant 200720I038 and Spanish MINECO (AGL2013-48756-R) to Bruno Contreras-Moreira.