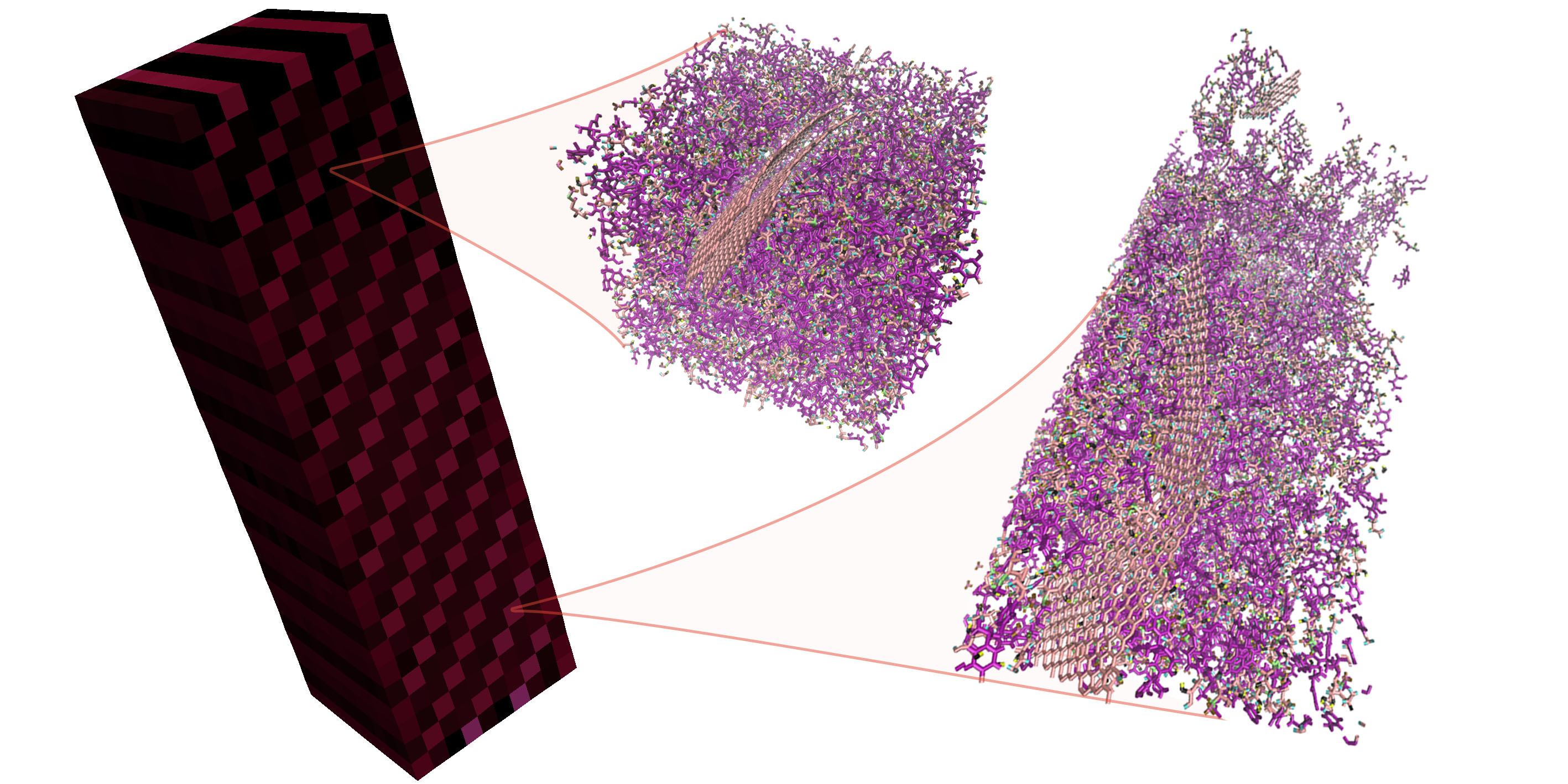
Heterogeneous Multiscale Method implementation featuring Deal.II (FE) and LAMMPS (MD). Enables simulations coupling semi-concurrently the evolution of an atomistic and a continuum system. The evolution of the continuum system drives the mechanical evolution of the periodic homogeneous atomistic replicas.
More details about this algorithm can be found in the following publication:
Maxime Vassaux, Robin Richardson and Peter Coveney. The heterogeneous multiscale method applied to inelastic polymer mechanics. Philosophical Transactions A, 377(2142), doi:10.1098/rsta.2018.0150.
Continuum mechanics equilibrium equations are solved on the basis of a linear elastic material. Non-linear stress/strain beahvior is captured running MD simulations of a sample of material subject to the continuum strain when needed.
The number of MD simulations can be drastically reduced through a graph reduction method with thresholding of "similarity" of one microstate's material history (strain vs time) with another - currently using L2 norm.
At the moment, there are strong dependencies on the versions of various packages required by this softare stack. The bootstrap/platform infrastructure below has been tested on a number of clusters/supercomputers (running linux) and is therefore recommended.
- gcc 4.9.2
- cmake 3.5.2
- python 3.0 or greater (if using graph-based clustering of MD simulations)
Deal.II needs to be compiled with the dependencies required to run the tutorial step-18, namely the following dependencies: MPI, PETSc (>3.6, 64bits), METIS (>4.0), MUMPS (>5.0), BOOST (>1.58), HDF5, LAPACK, MUPARSER, NETCDF, ZLIB, HDF5, and UMFPACK. Complete instructions can be found here.
cd /path/to/dealii-8.4.1/build/
cmake -DCMAKE_INSTALL_PREFIX=/path/to/install/dealii-8.4.1/ -DDEAL_II_WITH_MPI=ON -DDEAL_II_WITH_PETSC=ON ..
make install
make testLAMMPS version 17Nov16 has been tested and works correctly, more recent version can probably work as well with the LAMMPS scripts embedded in SCEMa. LAMMPS need to be compiled as a shared library with MPI support, along with the RIGID and USER-REAXC packages. Here is an example make invocation for the 17Nov16 version
cd /path/to/lammps-17Nov16/src
make yes-RIGID
make yes-USER-REAXC
make mode=shlib mpiAfter installing separately LAMMPS and Deal.II, and building your MD input lammps data file. Prepare the building directory:
cd /path/to/SCEMa
cp CMakeLists/example_machine.CMakeLists.txt CMakeLists.txt
mkdir buildThe file CMakeLists.txt needs to be edited to point toward the right installation path for Deal.II and LAMMPS. Then SCEMa executable can be compiled:
cmake ../
make dealammpsThe directory where the simulation is executed should contain at least the following data:
ll /path/to/simulation
... inputs_testname.json
... lammps_scripts_ffname -> /path/to/SCEMa/lammps_scripts_opls
... macroscale_input
... nanoscale_input
... clustering -> /path/to/SCEMa/clustering # when using clustering algorithm to determine and eliminate redundant molecular simulations
... surrogate_model -> /path/to/SCEMa/surrogate_model # when using a surrogate for molecular simulations ("stress computation method: 2")Most, if not all, of the simulation parameters are found in the configuration file inputs_testname.json:
cat /path/to/simulation/inputs_testname.json
{
"problem type":{
"class": "testname",
"strain rate": 0.002
},
"scale-bridging":{
"stress computation method": 0,
"approximate md with hookes law": 0,
"use pjm scheduler": 0
},
"continuum time":{
"timestep length": 5.0e-7,
"start timestep": 1,
"end timestep": 10
},
"continuum mesh":{
"fe degree": 1,
"quadrature formula": 2,
"input": {
"style" : "cuboid",
"x length" : 0.03,
"y length" : 0.03,
"z length" : 0.08,
"x cells" : 3,
"y cells" : 3,
"z cells" : 8
}
},
"model precision":{
"md":{
"min quadrature strain norm": 1.0e-10
},
"clustering":{
"spline points": 10,
"min steps": 5,
"diff threshold": 0.000001,
"scripts directory": "./clustering"
}
},
"molecular dynamics material":{
"number of replicas": 1,
"list of materials": ["matname"],
"distribution": {
"style": "uniform",
"proportions": [1.0]
},
"rotation common ground vector":[1.0, 0.0, 0.0]
},
"molecular dynamics parameters":{
"temperature": 300.0,
"timestep length": 2.0,
"strain rate": 1.0e-4,
"number of sampling steps": 100,
"scripts directory": "./lammps_scripts_ffname",
"force field": "ffname"
},
"computational resources":{
"machine cores per node": 24,
"maximum number of cores for FEM simulation": 1,
"minimum number of cores for MD simulation": 1
},
"output data":{
"checkpoint frequency": 1,
"visualisation output frequency": 1,
"analytics output frequency": 1,
"loaded boundary force output frequency": 1,
"homogenization output frequency": 1000
},
"directory structure":{
"macroscale input": "./macroscale_input",
"nanoscale input": "./nanoscale_input",
"macroscale output": "./macroscale_output",
"nanoscale output": "./nanoscale_output",
"macroscale restart": "./macroscale_restart",
"nanoscale restart": "./nanoscale_restart",
"macroscale log": "./macroscale_log",
"nanoscale log": "none"
}
}The data files for each replica of the tested molecular structure need to be prepared using the init_materials executable, and positioned in ./nanoscale_input (this path can be modified in the configuration file inputs_testname.json). If two replicas of the material matname are used, it should contain the following data:
ll /path/to/simulation/nanoscale_input
... matname_1.data
... matname_1.json
... matname_2.data
... matname_2.json
... init.matname_1.bin
... init.matname_1.length
... init.matname_1.stiff
... init.matname_1.stress
... init.matname_2.bin
... init.matname_2.length
... init.matname_2.stiff
... init.matname_2.stressMore complex finite element meshes for the continuum scale (than simple rectangular parallelepiped) can be simulated. Simply, a GMSH mesh file needs to be placed in ./macroscale_input.
Finally, the simulation can be run:
mpiexec /path/to/SCEMa/dealammps inputs_testname.jsonTo restart from a previous simulation, checkpoint files stored in ./nanoscale_restart and ./macroscale_restart must be placed, respectively, in ./nanoscale_input/restart and ./macroscale_input/restart.
Vassaux, M., Richardson, R. A., & Coveney, P. V. (2019). The heterogeneous multiscale method applied to inelastic polymer mechanics. Philosophical Transactions of the Royal Society A, 377(2142), 20180150.
Vassaux, M., Sinclair, R. C., Richardson, R. A., Suter, J. L., & Coveney, P. V. (2019). The Role of Graphene in Enhancing the Material Properties of Thermosetting Polymers. Advanced Theory and Simulations, 2(5), 1800168.
Vassaux, M., Sinclair, R. C., Richardson, R. A., Suter, J. L., & Coveney, P. V. (2020). Towards high fidelity materials property prediction from multiscale modelling and simulation, Advanced Theory and Simulations, 3(1), 1900122.