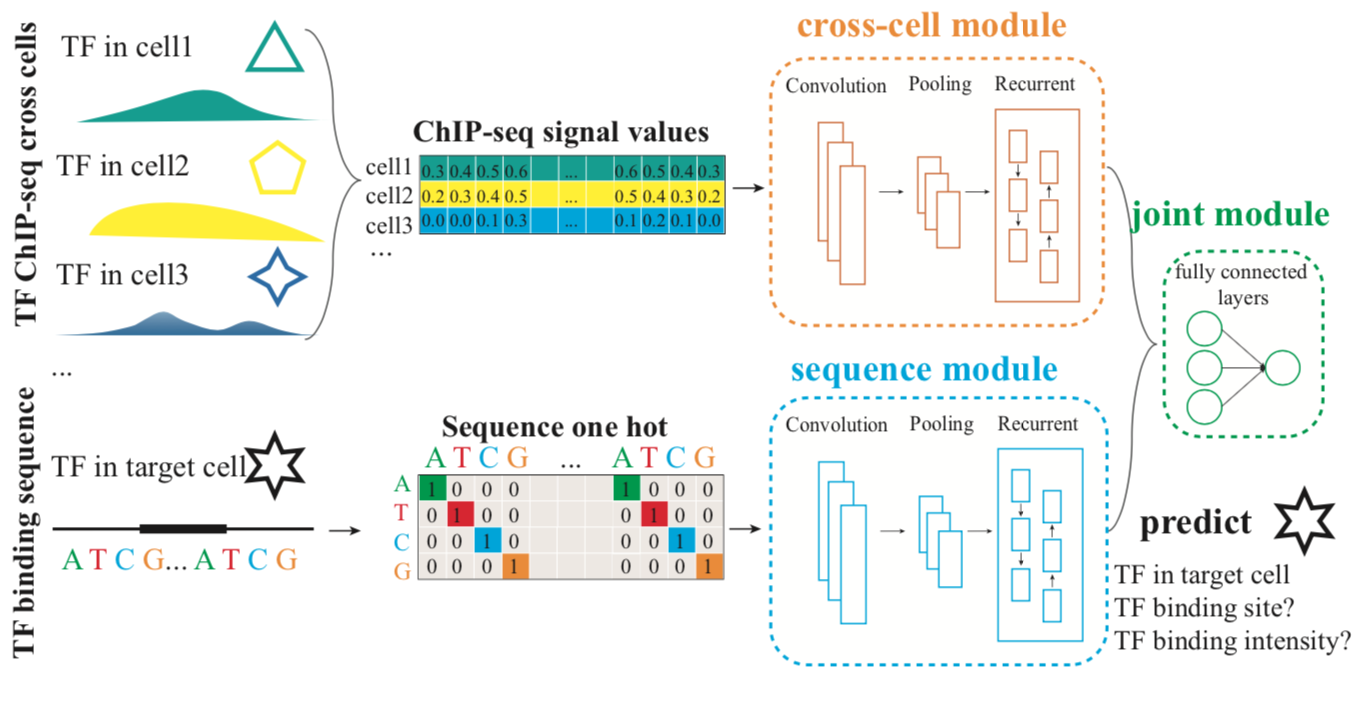
This is the implementation code of DeepBSI, a multimodal deep learning framework for predicting the transcription factor binding site and intensity.
Python 3.8 or higher
pybedtools 0.8.2
pyfasta 0.5.2
pyBigWig 0.3.18
tensorflow 2.4.1
keras 2.4.3
- The TF ChIP-seq datasets
The 10 TF ChIP-seq data cross cells were all downloaded from ENCODE. You can download from ENCODE through the ID directly. We provide all of the links in file data/datasets_links.txt. To train the model, we put the data of TF CTCF in cells A549 and GM12878 in the data folder.
| TF | cell | type | links |
|---|---|---|---|
| CTCF | A549 | broadPeak | https://www.encodeproject.org/files/ENCFF001XLL/@@download/ENCFF001XLL.bed.gz |
| CTCF | A549 | broadPeak | https://www.encodeproject.org/files/ENCFF001XLN/@@download/ENCFF001XLN.bed.gz |
| CTCF | A549 | narrowPeak | https://www.encodeproject.org/files/ENCFF002DBU/@@download/ENCFF002DBU.bed.gz |
| CTCF | A549 | bigWig | https://www.encodeproject.org/files/ENCFF413SFF/@@download/ENCFF413SFF.bigWig |
| CTCF | GM12878 | bigWig | https://www.encodeproject.org/files/ENCFF886KRA/@@download/ENCFF886KRA.bigWig |
- The reference genome
The human reference genome and their sizes of each chromosomes can be downloaded from UCSC (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/). We only provide three chromosomes (ch19, chr21, chr22) in the data folder.
python deepBSI_binding_site.py
python deeBSI_binding_intensity.py
The arguments of two scripts are the same, and some of the them are listed here.
| Argument | Default | Description |
|---|---|---|
| tf_name | CTCF | The TF you are interest. |
| target_cell | A549 | The cell you are interest. |
| cross_cells | GM12878 | The ChIP-seq data in other cells of the same TF. |
| train_chroms | chr19 | The chroms used to train (chromX and chrom1-22) |
| valid_chroms | chr22 | The chroms used to valid (chromX and chrom1-22) |
| test_chroms | chr21 | The chroms used to test in general use(chromX and chrom1-22) |
| data_dir | ../data/ | The fold contain TF ChIP-seq data. (1) Narrow peak(bed). (2) broad peak(bed) and signal |
| output_dir | ../DeepBSI_output_binding_site/ | The output dir. |
| ref_genome_fa | ../genomes/hg19.fa | The reference genome of human. |
| ref_genome_size | ../genomes/hg19.chrom.sizes | The size of human reference genome.) |
Please cite our work if you find our code/paper is useful to your work.
@article{Zhang,
title={DeepBSI: a multimodal deep learning framework for predicting the transcription factor binding site and intensity},
author={Peng Zhang, Shikui Tu},
journal={BIBM},
volume={},
number={},
year={2021},
month={},
pages={}
}