
Chrom3D is a computational framework for efficient reconstruction of 3D genome structures using chromosome contact data (Hi-C, TCC and 5C data) and optionally lamin ChIP-seq data.
Chrom3D is tested on MacOS & Linux. If you try to install it under Windows, please send us a few words on how it went.
Chrom3D is dependent on the boost library (>=1.54). If you do not have it, you can install it using (e.g. on Ubuntu):
sudo apt-get install libboost-dev
Otherwise, go to http://www.boost.org/users/download/
If you have boost installed, but still have installation problems when running make, find the boost directory using following command:
whereis boost
Then add the boost path to the Makefile:
all:
g++ -I [insert path from 'whereis boost' here] -I $(TCLAPINCLUDE) $(SRCPATH)/Util.cpp $(SRCPATH)/Bead.cpp $(SRCPATH)/Chromosome.cpp $(SRCPATH)/Randomizer.cpp $(SRCPATH)/Constraint.cpp $(SRCPATH)/Model.cpp $(SRCPATH)/MCMC.cpp $(SRCPATH)/Chrom3D.cpp -o Chrom3D
Then, make should work.
The final structures from Chrom3D are saved in "cmm" format that can be visualized in Chimera (https://www.cgl.ucsf.edu/chimera/). R programming language (https://www.r-project.org/) can also be used to visualize the structures and preform statistical calculations, but this requires customized code (we provide examples below).
Download Chrom3D source package here: https://github.com/Chrom3D/Chrom3D/releases/tag/v1.0.2
Enter Chrom3D directory and compile the program by typing make in the terminal.
The resulting Chrom3D binary should then be created for you in this folder. If you want Chrom3D available in your path, you can move the binary to somewhere available in your $PATH, for example ~/bin/.
Errors? Please report them under Issues
To make sure that Chrom3D is working properly on your machine:
./Chrom3D --version
The output should be as following:
./Chrom3D version: 1.0.2
-
As input file, we will use toy-global-example.gtrack file found in Chrom3D/test_files directory consisting of 2x2 (diploid cell) chromosomes with information on pairwise chromosomal interactions and interactions with lamin-B-proteins
-
To reconstruct this toy-genome, run following command (explanation below):
./Chrom3D -o ./test_files/toy-global-example.cmm -r 3.0 -n 50000 -l 5000 --nucleus ./test_files/toy-global-example.gtrack
-o ./test_files/toy-global-example.cmmgives instructions to save the final output structure in cmm format in Chrom3D/test_files directory. The size of the nucleus (3.0 µm) is set using-r 3.0. Using--nucleus, the model will be constrained within the nucleus. The-n 50000parameter sets the number of iterations, and-l 5000specifies that logging information should be output every 5000 iteration. The input file is specified using "./test_files/toy-global-example.gtrack".
- The output from this command should look like this:
# chromosomes: 4
# beads: 400
# interactions: 270
# interactions with given weight: 0
# non-bounded interactions with given distance: 0
# upper-bounded interactions with given distance: 0
# lower-bounded interactions with given distance: 0
# periphery beads: 120
# periphery beads (with weights): 0
# center beads: 280
# center beads (with weights): 0
- Use Chimera to visualize the final structure:
<chimera_install_dir>/UCSF-Chimera64-1.10.2/bin/chimera ./test_files/toy-global-example.cmm
- A tip: To visualize the transparent nuclear boundary in Chimera, go to "Favorites/Command Line" and in the command line window (bottom), write following command:
shape sphere center 0,0,0 radius 3.0 color #FFE4B5
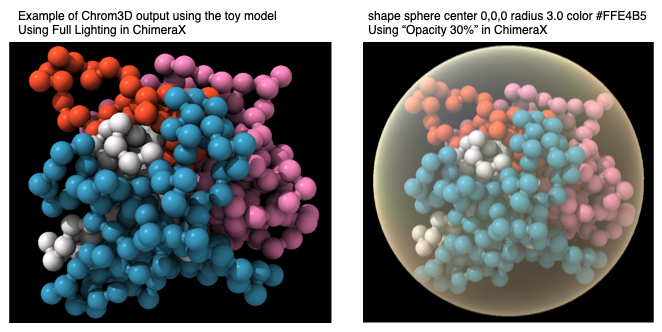
Note: The output structure may differ to a small degree due to different versions of random number generator
The chromosome/bead moves used by Chrom3D are illustrated below:

The default value of temperature T parameter is 1 and cooling rate c is 0. This means that the default optimization is based on Metropolis-Hastings algorithm since T does not influence the acceptance probability of the “bad” move. The default setting is optimal for full-genome optimization and other complex systems. If you wish to use a simulated annealing approach, you need to set the value of the cooling rate c between 0 and 1. The temperature T parameter will decrease gradually during the simulation with the slope determined by c (Tnew = Tprevious*(1-c) for each accepted move. The idea behind cooling down the temperature during optimization is to speed up the convergence a the end of the simulation. It is, however, difficult to determine which cooling rate is appropriate to use and we recommend trying out different values. The rate will be dependent on the total number of iterations you wish to run during simulation and how steep you wish to narrow down the search. A simulated annealing approach usually works best in simple systems consisting of one or a few chromosomes. We recommend to have a smooth decrease of T, obtained by setting the cooling rate to c=5/n, where n is the total number of iterations.