A pipeline to study intratumor heterogeneity (ITH) with Canopy[1].
This pipeline has been inspired from the Marathon pipeline[2] proposed by the authors of Canopy. Marathon is a description of a conceptual pipeline using Falcon and Canopy. It is not a functional and automated pipeline. The goal of the ITH pipeline is to propose a working and automated pipeline.
In this documentation, patient ID has been replaced with ##, and tumors ID has been replaced with T1 and T2.
- tool : BWA
- input : a BAM file
- output : a BAM file
- scripts : scripts/cobalt/template_postalt.sh, scripts/cobalt/launch_postalt.sh
- tool : Platypus
- inputs : a normal BAM file, human genome reference file, regions file
- output : a normal VCF file
- scripts : scripts/cobalt/template_platypus.sh, scripts/cobalt/launch_platypus.sh
Then the VCF output file has been filtered on PASS value : scripts/keep_pass.sh.
- tool : Strelka2
- inputs : a tumor BAM file, its associated normal BAM file, human genome reference file, regions file
- output : a tumor VCF file
- scripts : scripts/cobalt/template_strelka2.sh, scripts/cobalt/launch_strelka2.sh
Then the VCF output file has been filtered on PASS value : scripts/keep_pass.sh.
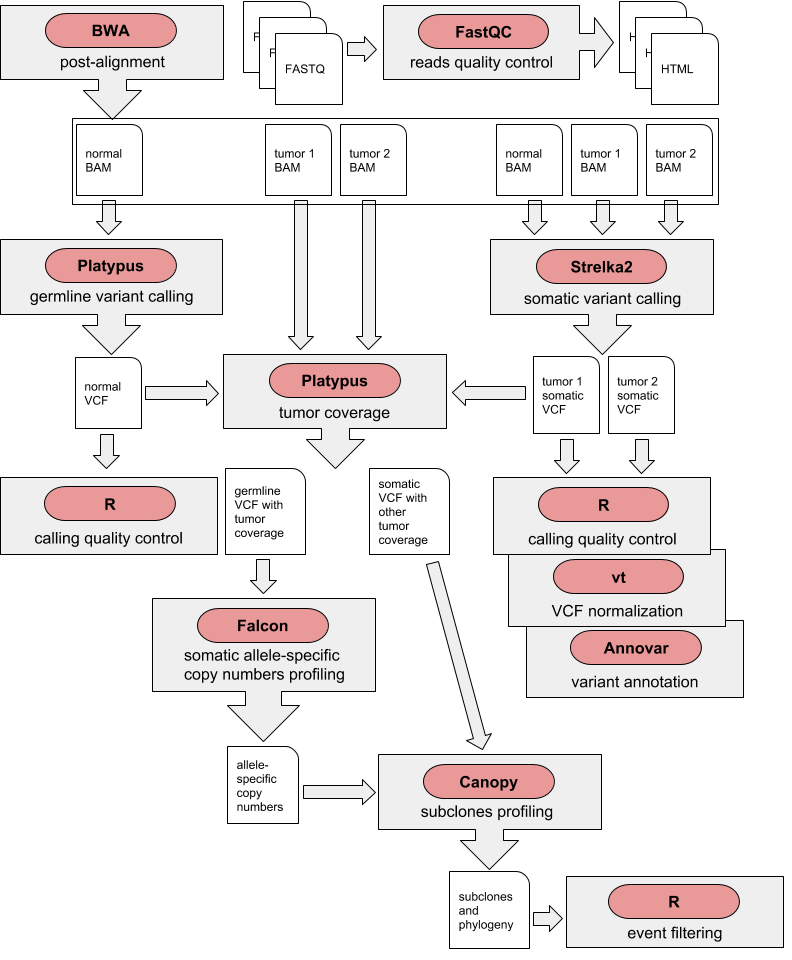
To generate four different charts :
| Germline AF distribution | Somatic Venn |
|---|---|
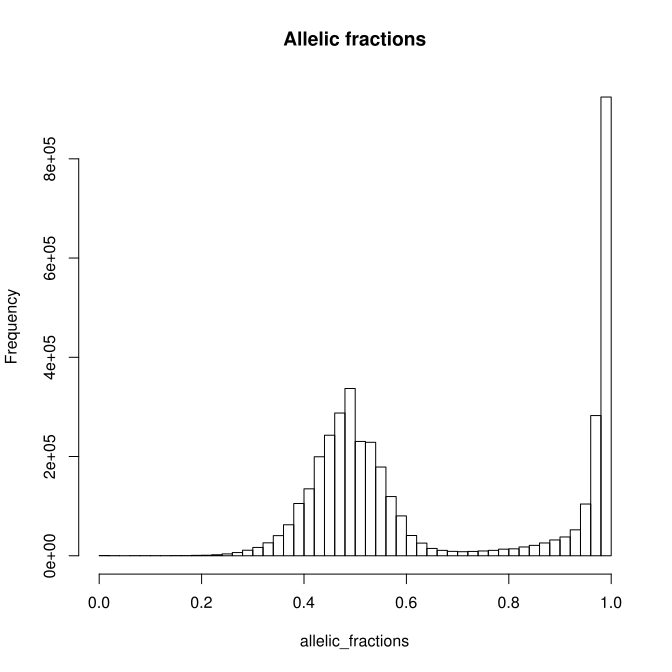 |
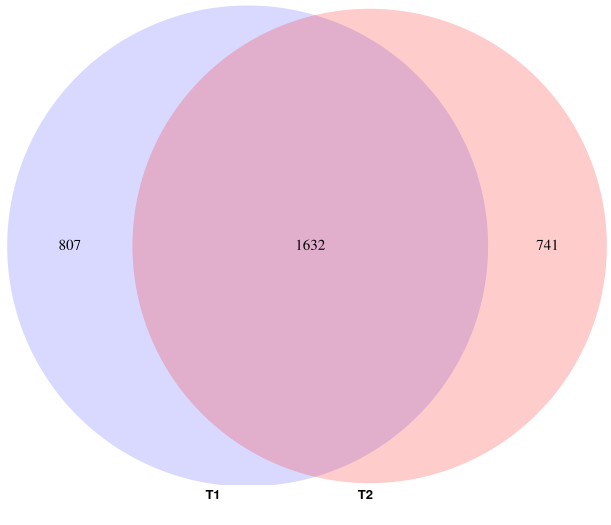 |
| Somatic AF distributions and overlap | Somatic / Germline overlap |
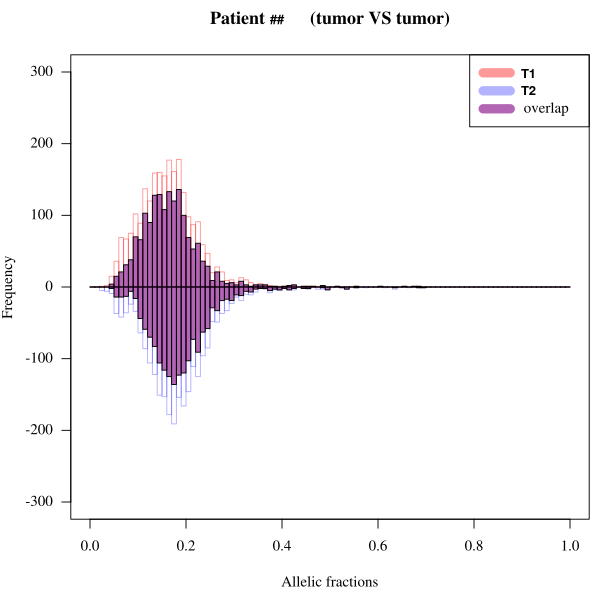 |
 |
- tool : R
- inputs : VCF files
- outputs : SVG files
- scripts : R_QC/*
The VCF can be normalized to have a format compatible with annovar.
- Platypus : mandatory
- Strelka2 : already compatible
- tool : vt
- inputs : a tumor VCF file
- output : a tumor VCF file
- script : scripts/normalize_vcf.sh
- tool : Annovar
- inputs : a VCF file
- output : a VCF file with extra columns
- script : scripts/run_annovar.sh
(In this pipeline, Annovar has been run on somatic VCF only)
For each patient, somatic calling of tumor1 & 2 give two variant lists with their respective positions and coverage. Germline calling also gives a variant list with its own positions and coverage.
We need the coverage of these positions in the others samples. For example, we need the coverage in tumor2 at the positions of tumor1 somatic variants. Inversely, we need the coverage in tumor1 at the positions of tumor2 somatic variants.
We also need tumor1 & 2 coverage at the positions of the germline variants.

- tool : Platypus
- inputs : a tumor BAM, a tumor VCF, human genome reference file, regions file
- output : a tumor VCF file
- script : scripts/calling_somatic_genotype/platypus_reads_*.sh
- tool : Platypus
- inputs : a tumor BAM, a normal VCF, human genome reference file, regions file
- output : a tumor VCF file
- script : scripts/calling_germline_genotype/platypus_genotype_*.sh
The script is parallelized by chromosome.
To split germline VCF by chromosome, use this script : scripts/split_vcf_chromosome.sh
- tool : Falcon (R package)
- inputs :
- args[1] = path_to_normal_VCF
- args[2] = patient_id
- args[3] = normal_sample_id
- args[4] = tumor1_sample_id
- args[5] = tumor2_sample_id
- args[6] = chr
- args[7] = path_to_output
- args[8] = path_to_lib_output
- args[9] = path_to_lib_qc
- outputs :
- a PDF with segmentation results
- a PDF with QC
- a TXT with copy numbers and their standard error
- a RDA with germline data loaded
- a RDA with tumor1 copy number computed
- a RDA with tumor2 copy number computed
- script : marathon/falcon/falcon.R
- example :
Rscript falcon.R /path/to/germline_VCF/splitted_by_chromosomes/sample.GERMLINE.chrY.vcf patient1 normal_sample_id tumor1_sample_id tumor2_sample_id Y /path/to/output/dir /path/to/marathon/libs/falcon.output.R /path/to/marathon/libs/falcon.qc.R
- tool : Falcon (R package)
- inputs :
- args[1] = tumor1 RDA file
- args[2] = path to Falcon TXT file containing coordinates (give a path like "/home/pgm/Workspace/MPM/marathon/falcon/output/patient_##/chr6/falcon.patient_##.tumor_placeholder.chr_6.output.txt" so it can take tumor1 and tumor2 TXT file thanks to the placeholder)
- args[3] = path to output directory
- args[4] = path to libs directory
- outputs :
- a TXT with the computed standard errors
- script : marathon/falcon/falcon_epsilon.R
It is important to note that these Falcon scripts use some custom libraries stored in marathon/libs/.
Sometimes, these libraries are simple overrides of Falcon with little modifications.
This step computes all input matrices required by Canopy, and performs SNA pre-clustering, and then a MCMC sampling to give subclones with composition and history.
This step has been parallelized by number of subclones.
- tool : Canopy (R package)
- inputs :
- args[1] = path to Falcon patient output directory
- args[2] = patient ID
- args[3] = tumor1 ID
- args[4] = tumor2 ID
- args[5] = path to tumor1 VCF file
- args[6] = path to tumor2 VCF file
- args[7] = path to tumor1 coverage VCF file at tumor2 positions
- args[8] = path to tumor2 coverage VCF file at tumor1 positions
- args[9] = path to output directory
- args[10] = number of subclones to generate
- args[11] = path to libs directory
- outputs :
- a BIC file with bayesian Information Criterion score for this number of subclones
- a SVG file with optimal number of clusters (non-informative without a comparison with others numbers of clusters)
- a PDF file with optimal number of subclones (non-informative without a comparison with others numbers of subclones)
- a PDF file with the likelihood of this number of subclones
- a RDA file containing the pre-clustering and the MCMC sampling computed for this number of subclones
- script : marathon/canopy/canopy.R
- tool : Canopy (R package)
- inputs :
- args[1] = patient ID
- args[2] = path to canopy patient output
- outputs :
- a SVG file with a plot of BIC of each number of subclones (to choose the better BIC)
- a PDF file with the visual generated tree
- a TXT file with the composition of each subclones (SNAs and CNAs)
- script : marathon/canopy/canopy_tree.R
patient_id = args[1] data_path = args[2] file_name = args[3] input_somatic_VCF_t1 = args[4] input_somatic_VCF_t2 = args[5] only_exonic = args[6]
- tool : Canopy (R package)
- inputs :
- args[1] = patient ID
- args[2] = path to canopy patient output
- args[3] = name of the TXT subclones composition file
- args[4] = path to the tumor1 VCF file
- args[5] = path to the tumor2 VCF file
- args[6] = 1/0. If 1, keep exonic SNAs only
- outputs :
- a TSV file with filtered events
- script : marathon/canopy/canopy_subclones_composition.R
It is important to note that these Canopy scripts use some custom libraries stored in marathon/libs/.
Sometimes, these libraries are simple overrides of Canopy with little modifications.
[1] Assessing intratumor heterogeneity and tracking longitudinal and spatial clonal evolutionary history by next-generation sequencing
Jiang, Y., Qiu, Y., Minn, A.J. and Zhang, N.R., 2016.
Proceedings of the National Academy of Sciences.
http://www.pnas.org/content/pnas/113/37/E5528.full.pdf
[2] Integrative pipeline for profiling DNA copy number and inferring tumor phylogeny
Eugene Urrutia Hao Chen Zilu Zhou Nancy R Zhang Yuchao Jiang
Bioinformatics, bty057 (2018)
https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/bty057/4838234?redirectedFrom=fulltext