This 'MultiGame' is comprised of two games: MatchTwo and SimpleSudoku, in order to compile the game you need to install Opentk which is located at http://www.opentk.com/ After installing Opentk you can open and edit the solution in Visual Studio
If you just want to play the Game you can download the basic zip file SmallRelease.zip located at https://github.com/K5rovski/MultiGame/tree/master/Multigame/MultiGame/bin
The look of the startscreen is :
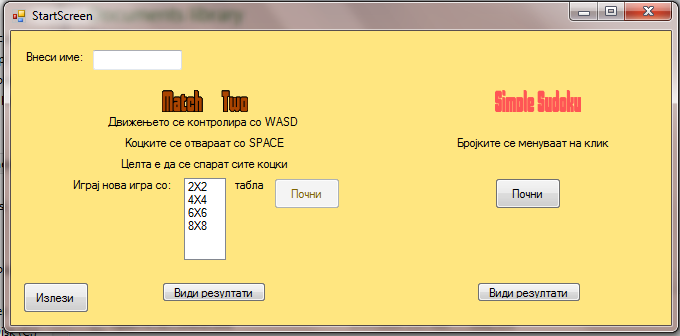
At the bottom of the window you can see two buttons which allow you to see the highscores of the two available games. If you haven't finished a game your Score is logged as Unfinished.
There are only 30 Spots for HighScores for the two games.
After each playing session the highscores are saved locally (in the file SavedScores.savedS).
After you enter your name you can play either the game MatchTwo or SimpleSudoku.
This is an example 3d game utilizing the OpenTK Library, the focus of the game is to match all the same cubes.
This is the look of the game:
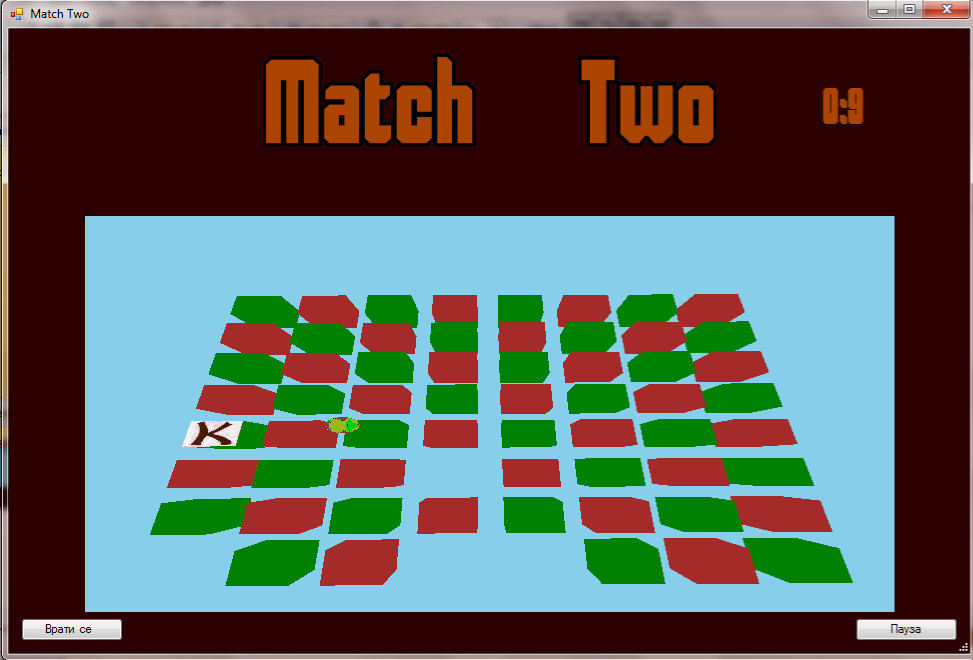
In order to play the game you must first enter a size of the board of cubes in the StartScreen.
The controls are as described in the starstcreen, wasd for movement and space to open a box, after you open two same boxes they are deleted and you need to match more boxes.
You can pause or go back to the StartScreen at any time.
After you match all the boxes your score is displayed bellow the title like so:
.png)
Your Score is based on the time passed since starting the game and the size of the board.
This is a simple implementation of Sudoku with moderate difficulty in which you control input by clicking on an empty square and then choosing a number to enter into the empty spot.
If you enter a wrong number the square will turn red.
This is the look of the game:
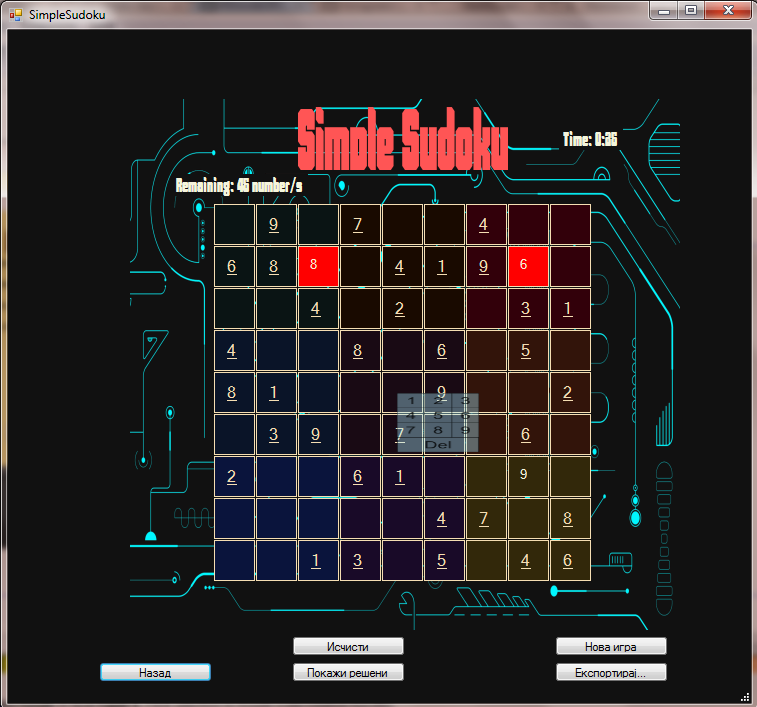
There are several standard options such as clearing the board, starting a new game or showing the solved board, also you can export several sudoku boards in an image file on your computer.
Your Score is based on the time passed since starting the game.
'public void Keyboard_In (int kliknat)' is the function in MatchTwo/Tabla.cs that is called whenever a player presses SPACE, it deals with the (de)selection of the cubes.
The argument of the function is the location of the newly selected cube.
if (SelektiranOne == -1) {
SelektiranOne = kliknat;
tabla[SelektiranOne].ChangeDirection(); //
}
if the first selection is empty the code above sets it to the new location 'kliknat' and changes the direction of the selected cube
else if (SelektiranOne!=-1 && SelektiranTwo!=-1) {
tabla[SelektiranOne].ChangeDirection(); //
tabla[SelektiranTwo].ChangeDirection(); //
SelektiranTwo=SelektiranOne=-1;
SelektiranOne = kliknat;
tabla[SelektiranOne].ChangeDirection();
}if the two selections are not empty it changes their location, direction and sets the first selection to 'kliknat' changing its direction and deselects the second selection.
else if (SelektiranOne == kliknat) {
tabla[SelektiranOne].ChangeDirection(); // odselektiraj krajna pozicija i nasoka
SelektiranOne=-1;
}if you select the already selected element it deselects it and changes the direction.
else if (SelektiranTwo==-1){
SelektiranTwo=kliknat;
tabla[SelektiranTwo].ChangeDirection(); // selektiraj
CheckDelete(SelektiranOne,SelektiranTwo); // ako se ednakvi teksturite napravi selecttwo i izbrisi two
}if only the first selection is made this sets the second selection,changes its direction, also checking to see if the two selections are the same.
// Kvadrat.cs
public void ChangeDirection() {
KrajnaPozicija = opseg - KrajnaPozicija;
moving = true;
if (nasoka == -1)
{
nasoka = 1;
}
else {
nasoka = -1;
}
}this switches KrajnaPozicija between 0 and opseg, sets the moving flag and switches nasoka between -1 and 1.
// Tabla.cs
public void CheckDelete(int a, int b) {
if (tabla[a].N_Tekstura == tabla[b].N_Tekstura &&
tabla[a].Partner==-1 && tabla[b].Partner==-1) {
tabla[b].ToDelete=true; // delete flag true when finishes
tabla[b].Partner = a; // spari go so a
tabla[a].Partner=b;
SelektiranOne = SelektiranTwo = -1;
}
}
if the pictures on the two selected cubes are the same and both of them don't have a Parther so far, this sets the Todelete flag of the second selection, sets the partner of both cubes, and deselects them.
// Tabla.cs
public void Update()
{
int i = 0;
for (; i < tabla.Count; i++)
{
if (!tabla[i].Deleted && !tabla[i].moving && tabla[i].ToDelete)
{
if (tabla[i].Serenity == 20)
{
tabla[i].Deleted = true;
tabla[tabla[i].Partner].Deleted = true;
//Maybe Change RPlus();
}
break;
}
}
}this iterates through all of the cubes checking every cube if its not deleted, not moving and is slated to delete, and if the cube has been resting for 20 timer ticks , then it deletes it and its Partner.
The only posible Change is the call to RPlus which moves the selection ball to the right when a deletion occurs, this is currently commented because while playing the game it moves the ball unexpectedly, while now the ball will remain in an empty spot until the next hit of WASD when the game will resume normally.