Demo of LabKey Server's module file-based pipeline using RNASeq data. This simple pipeline reformats an existing matrix into one appropriate for importing into LabKey, performs further analysis and generates images, and imports the analyis results.
-
Install LabKey Server (14.3 is required) and ensure the Microarray module is included.
-
Install this module by zipping up the root RNASeqMatrixDemo directory and copying to the server's "modules" directory.
-
Install R and the "edgeR" bioconductor package.
- source("http://www.bioconductor.org/biocLite.R")
- biocLite("edgeR")
- Use the "data/GSE56845_counts.txt" RNASeq matrix or obtain the original "GSE56845_gene_counts_Rhesus_Ensembl.txt" from Geo: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE56845
Barrenas F, Palermo RE, Agricola B, Agy MB, Aicher L, Carter V, Flanary L, Green RR, McLain R, Li Q, Lu W, Murnane R, Peng X, Thomas MJ, Weiss JM, Anderson DM, Katze MG. Deep transcriptional sequencing of mucosal challenge compartment from rhesus macaques acutely infected with simian immunodeficiency virus implicates loss of cell adhesion preceding immune activation. J Virol. 2014 Jul 15;88(14):7962-72. doi: 10.1128/JVI.00543-14. Epub 2014 May 7. PubMed PMID:
- Start the server.
- Create a new assay folder, enable the RNASeqMatrixDemo module.
- Upload the "GSE56845_counts.txt" matrix file to the folder.
- Create a new ExpressionMatrix assay with the default fields.
- Add the "Feature Annotation Sets" webpart to the portal.
- Import the "illumina-feature-set.tsv" found in the "data" directory of this module.
- Create a new "General" assay named "DifferentialExpression" and with the following fields:
- Run Fields:
- multiDimensionalScalingPlot (label=MDS, type=File)
- meanVariancePlot (label=Variance, type=File)
- Data Fields:
- FeatureId (Text)
- logConc (Number)
- logFC (Number)
- pvalue (Number)
- padj (Number)
From the file-browser, select the matrix file then click "Import Data".
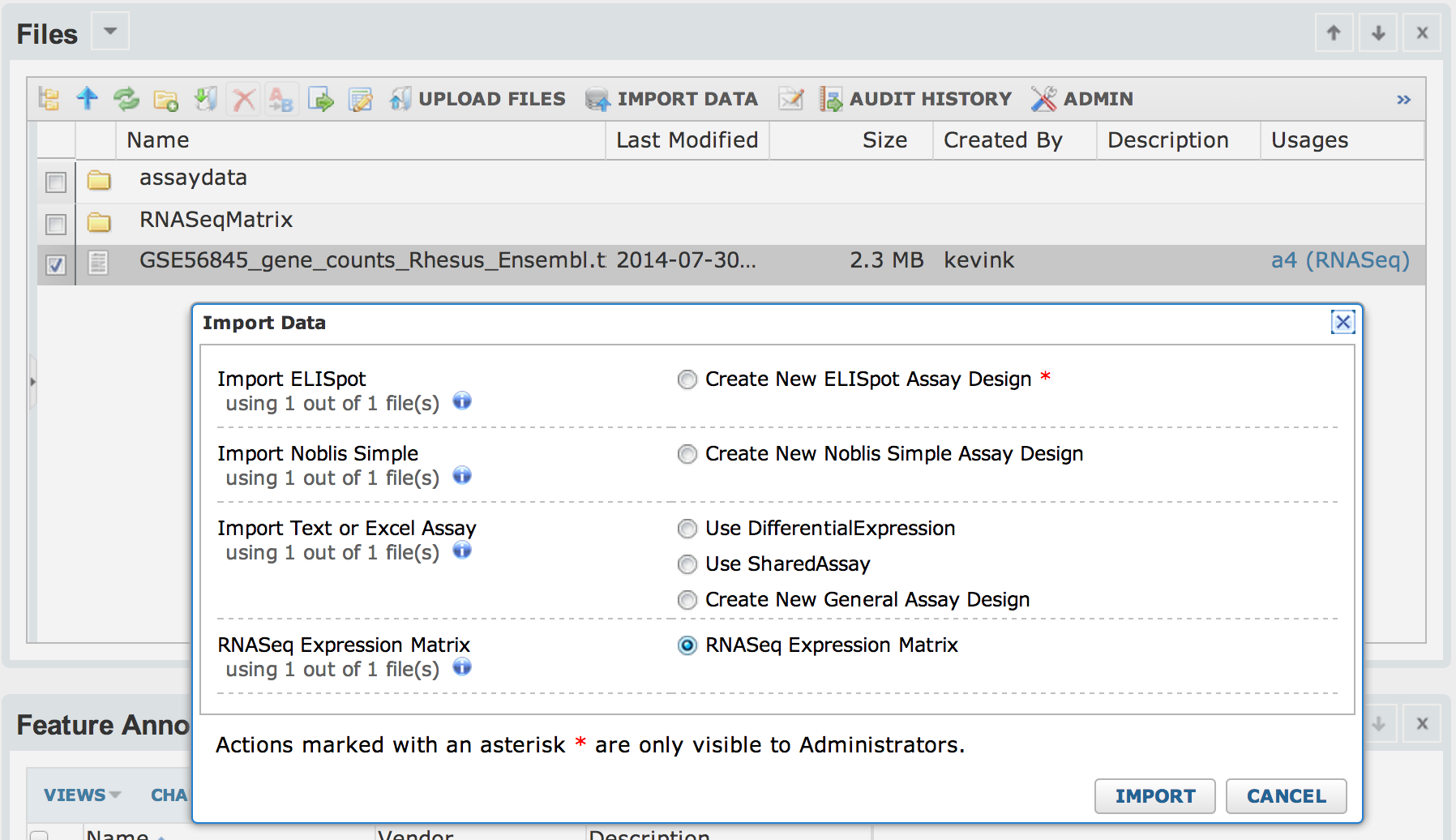
Next, enter a name for the run and select the target ExpressionMatrix assay and the feature annotation set you created during the setup. By default, the "Import Values" checkbox is unchecked and the matrix will not be imported into the assay -- only pointers to the original data file and the output files will be captured as part of the run.
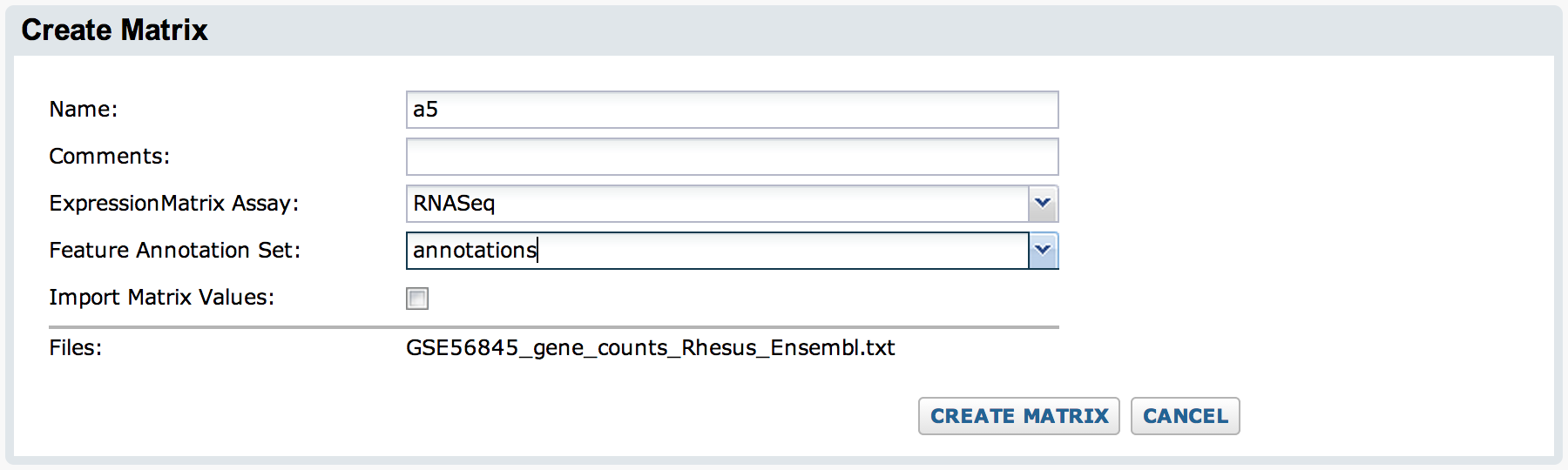
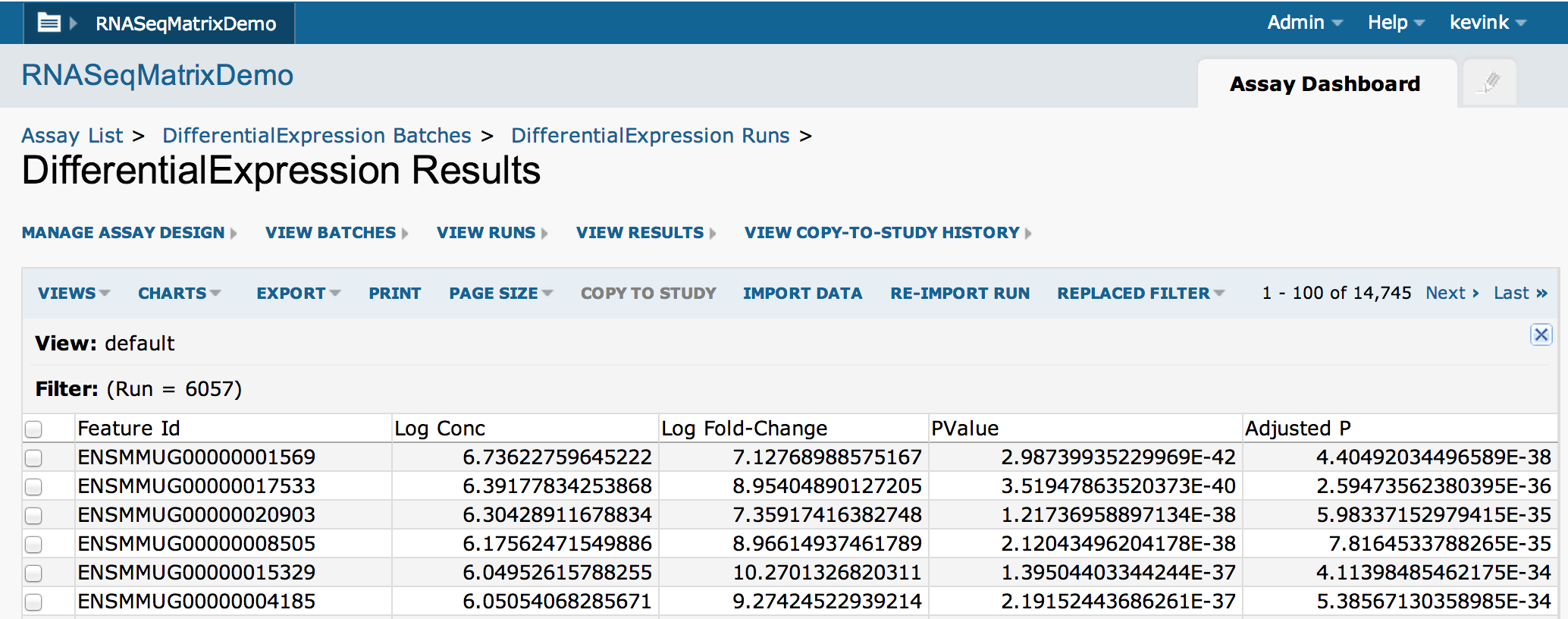
As the pipeline executes, files are written into the pipeline directory under a "RNASeqMatrixDemo" directory. The pipeline will create two assay runs: one ExpressionMatrix assay run and one DifferentialExpression assay run. As a part of the differential expression analysis, a series of images will also be generated. Further analysis could be performed on the values imported into the DifferentialExpression assay.
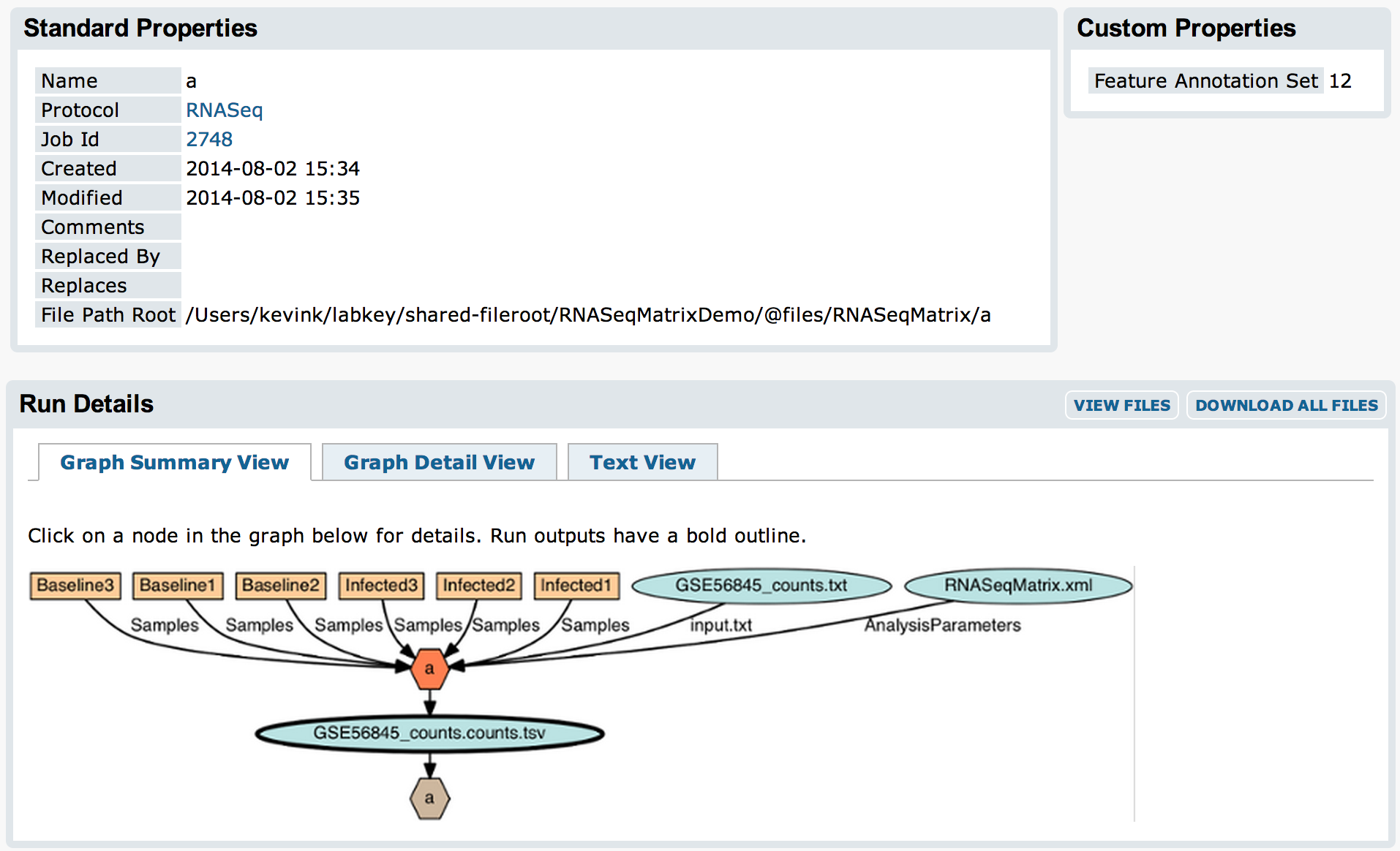
The original input file and samples in the filtered matrix will be attached to an experiment run for the ExpressionMatrix assay import:
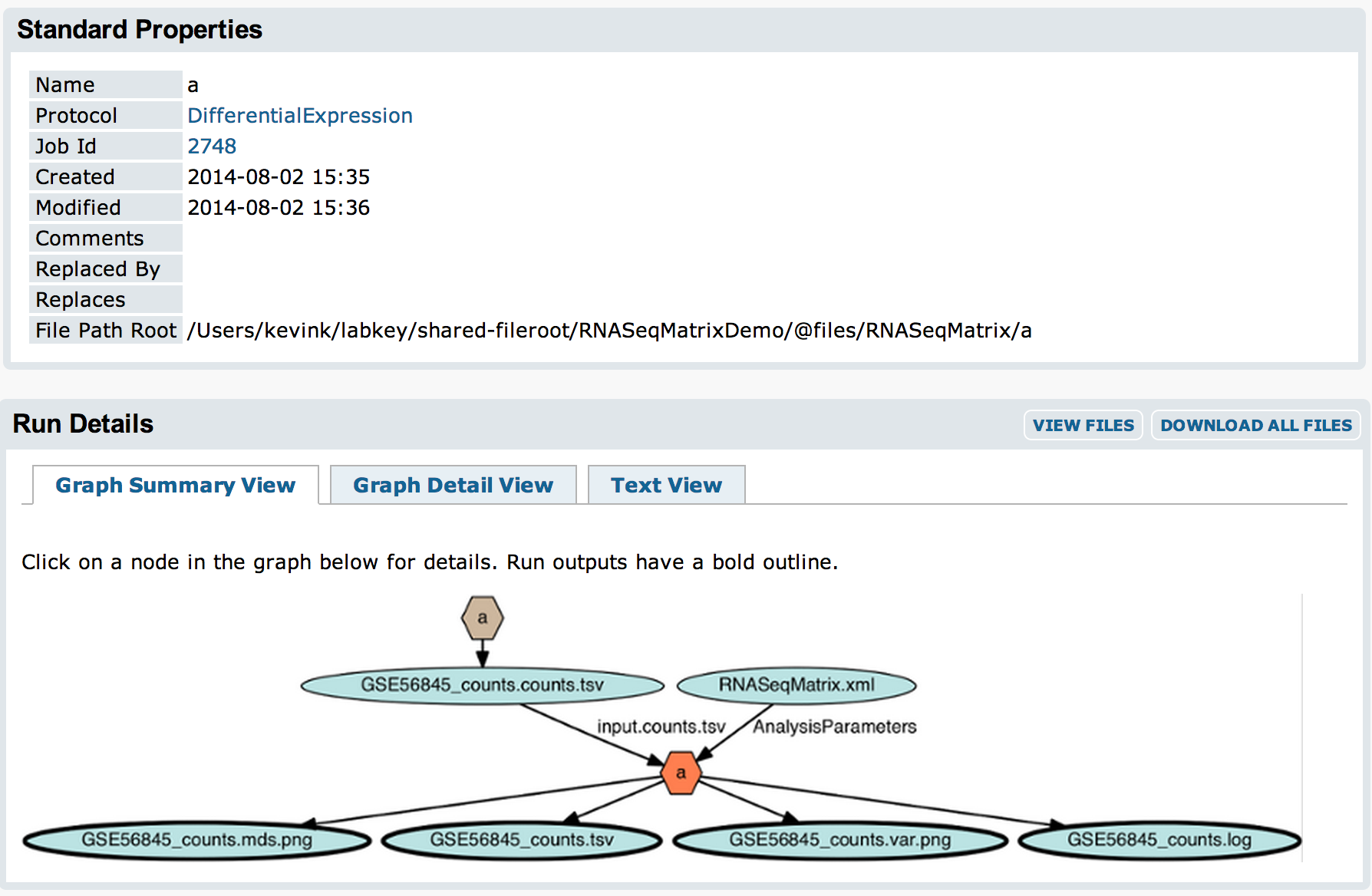
The filtered matrix file and the output files will be attached to an experiment run for the DifferentialExpression assay import:
The output file values will be imported into the assay's results table:
-
Issue 19066: Can't write task that has inputs and outputs of the same extension
-
Issue 19064: Can't write task that operates on full input file names instead of extension