[TOC]
This project contains all kind of papers that helps me a lot at work, for example, some help me to speed up the computation of TSP problem, some help me to overcome the GPS trajectory correction, GPS trajectory section, some help me to do a better jobs in Navigation(CNNs), some help me to recommend a better route in route planning problem(RNNs) and other many many jobs, I post them here wish to help someone who needed and so that he/she can make things more better.
You can clone and share it with others, also if you have some other papers/documents/books that you think they may help others, just push them.
Below are some details about the papers in the project.
-
genetic algorithm
The papers in genetic algorithm(GA) folder are mainly about TSP, and it helps me to speed up the computation and improve the accuracy of route planning. For example I implemented the GA in parallel and it was three to four times faster, left bottom is normal computation and right bottom is parallel computation in my local machine, and when I put it on the server, I run 10 threads parallel to calculate, and it can help me plan 40 points in 1-2 seconds.


And the algorithm seems to work very well in TSPLIB which can be download from https://www.iwr.uni-heidelberg.de/groups/comopt/software/TSPLIB95/ . Below I show some accuracy viewer of the route planning using GA.
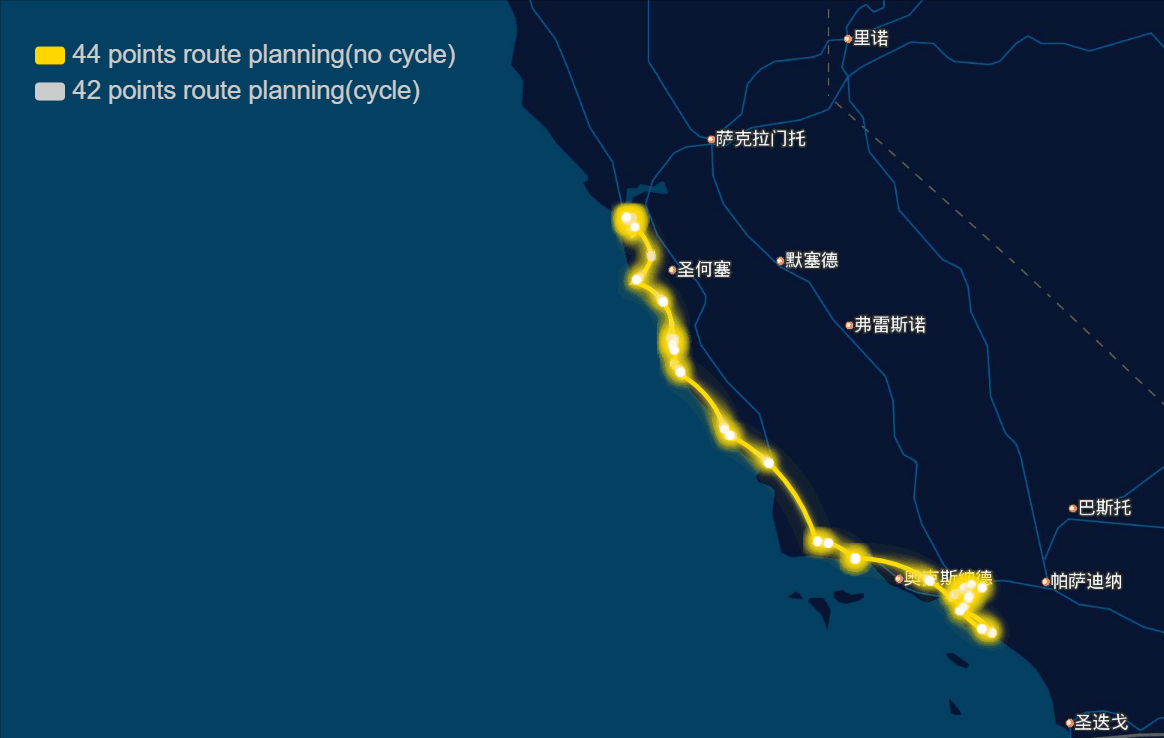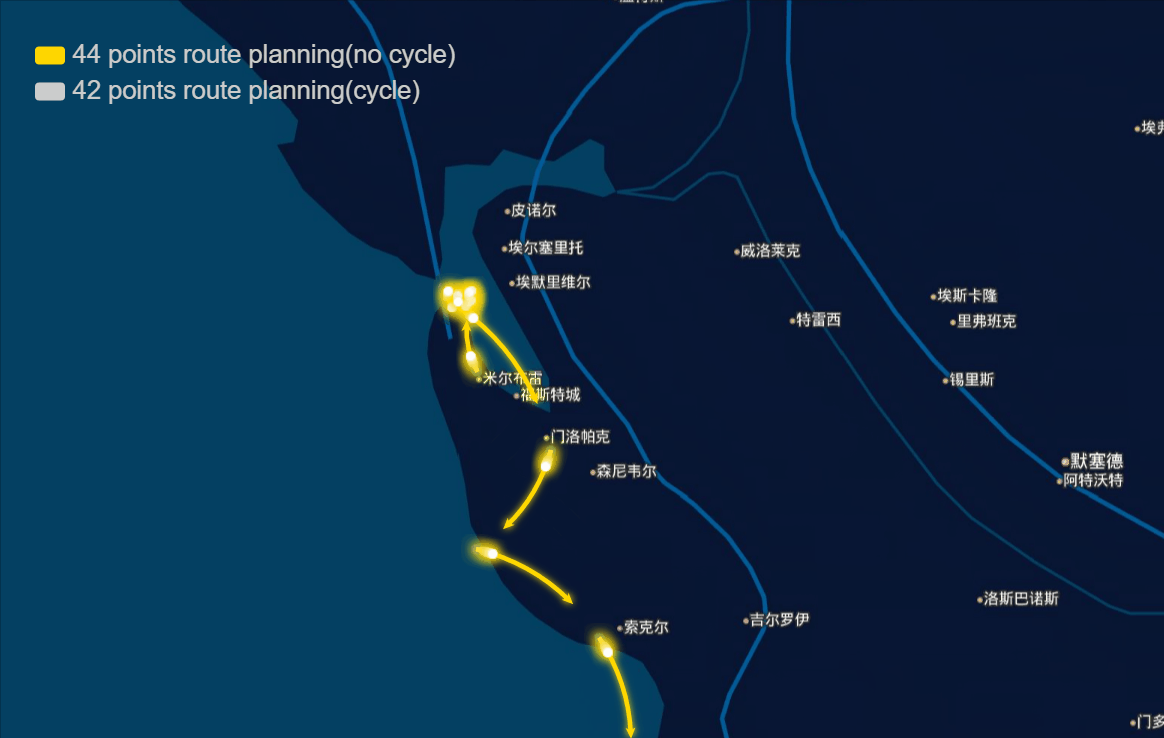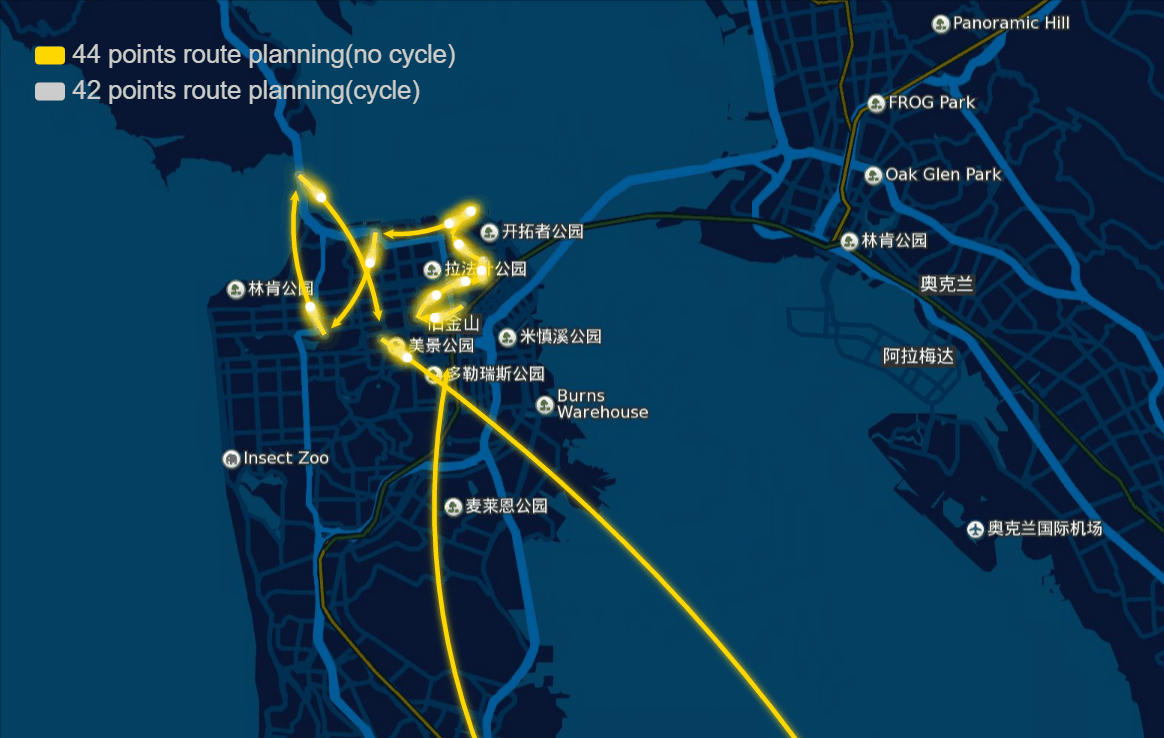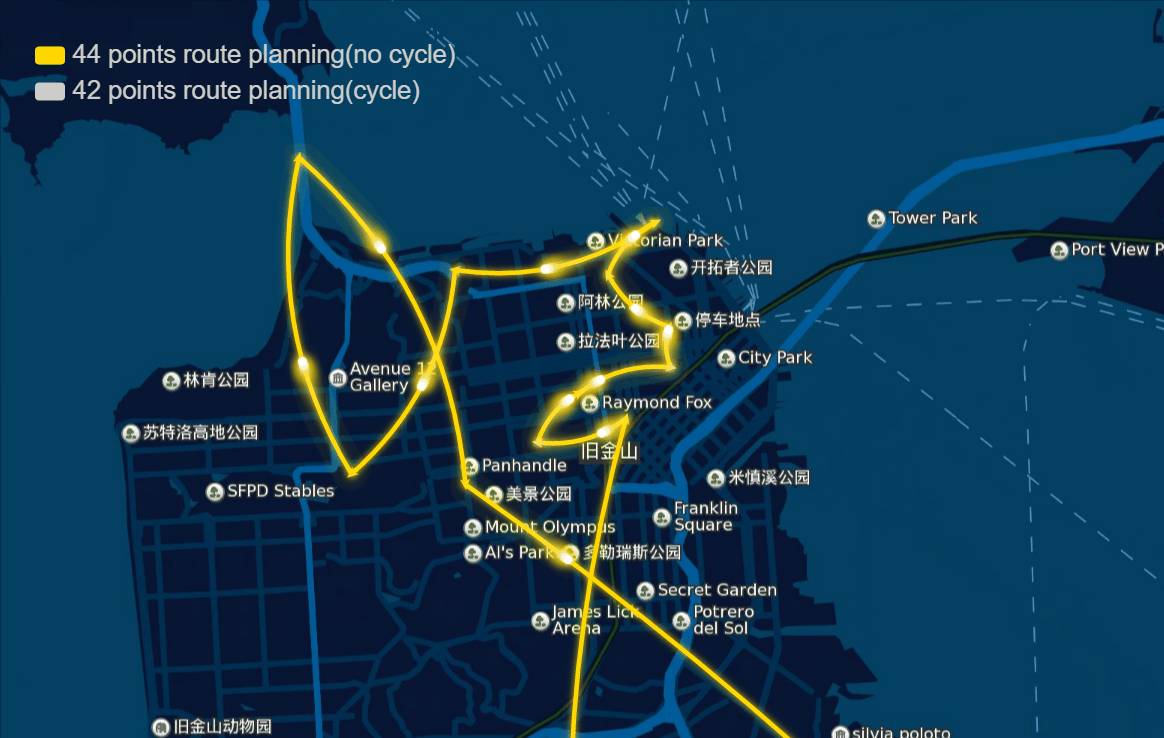
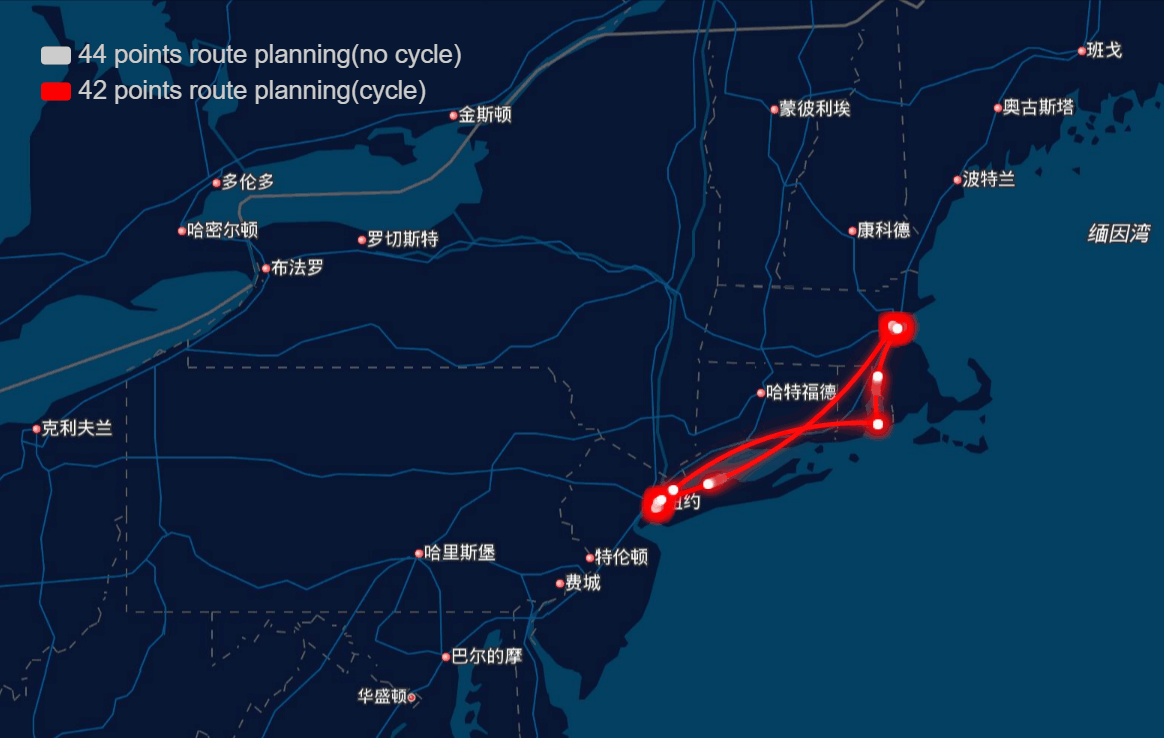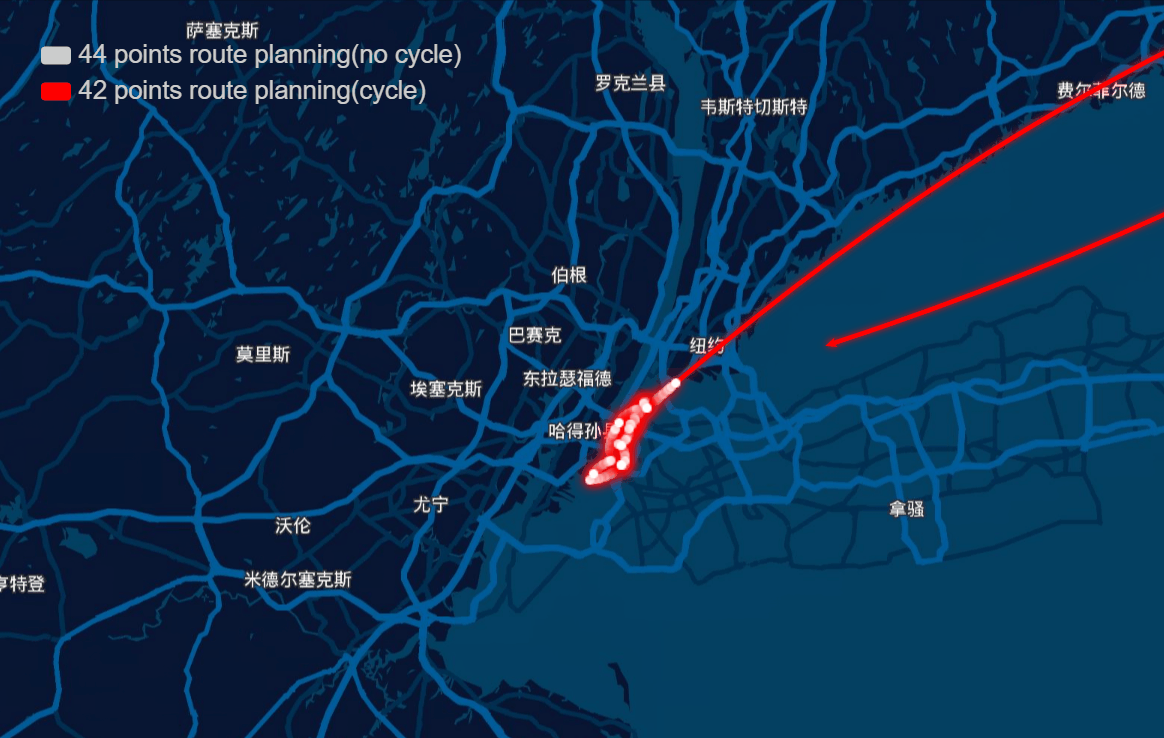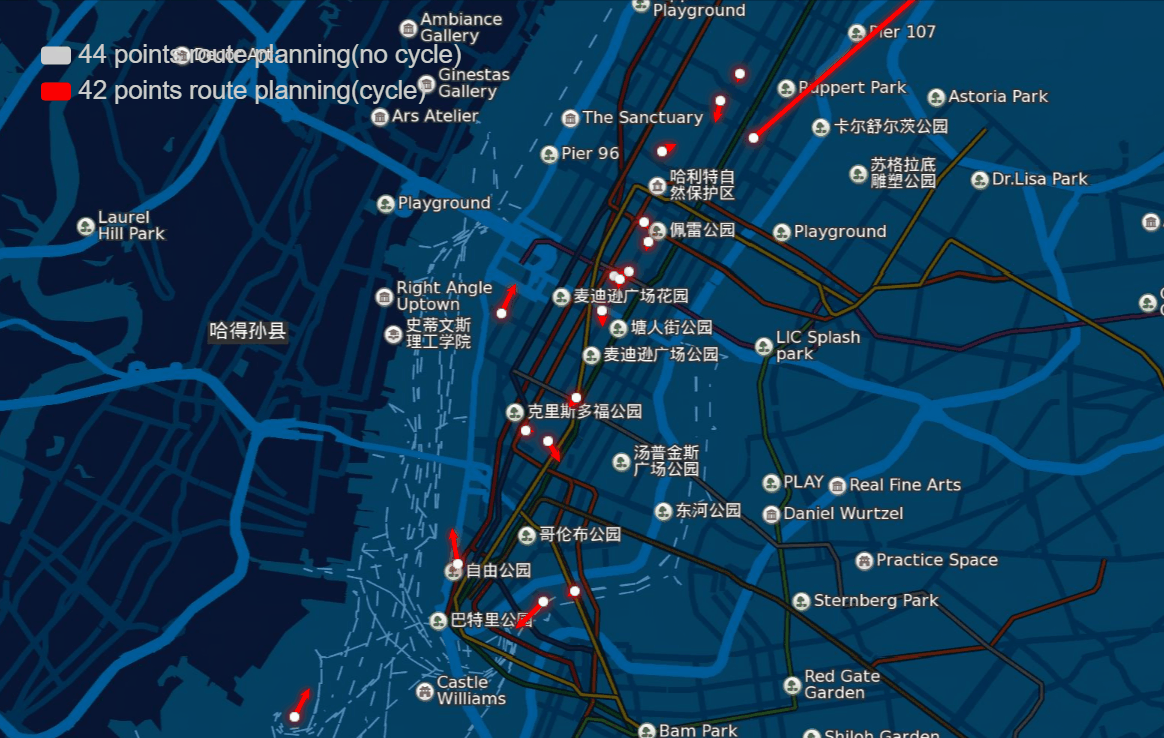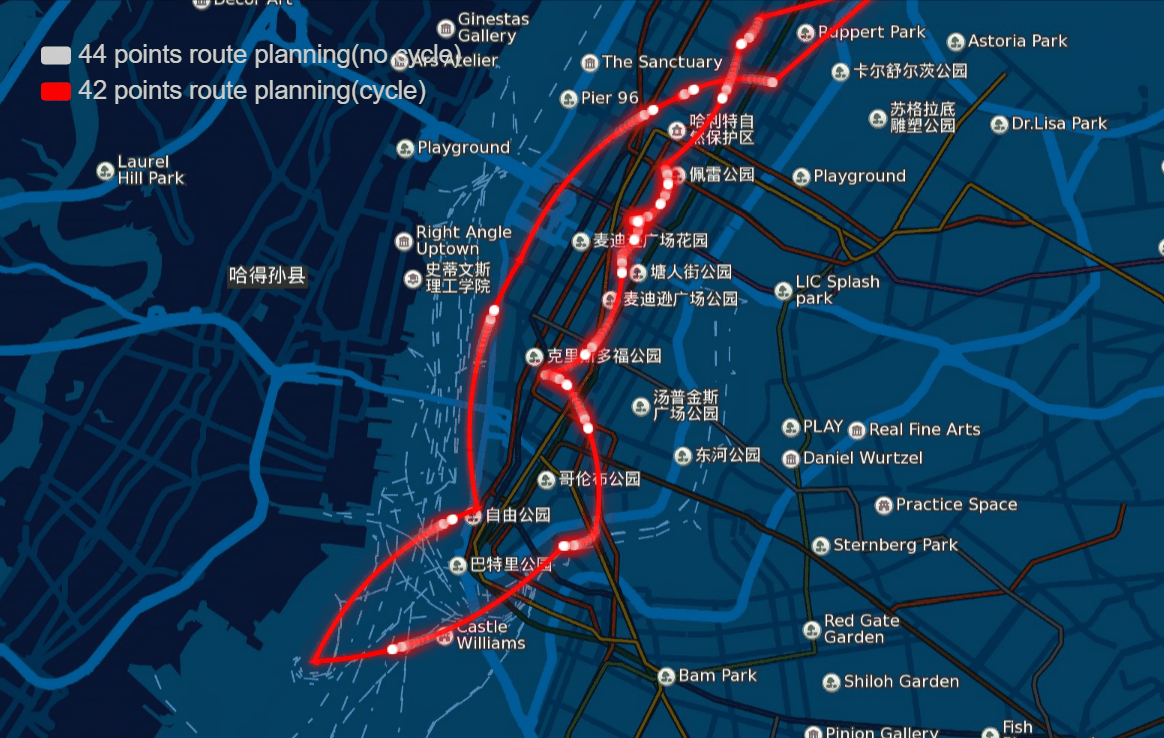
-
DBSCAN
The papers in DBSCAN folder are mainly about how to cut the GPS trajectory into moving or stay blocks. I implemented the DBSCAN algorithm and modified its search logistic so that the algorithm can search through time based points. Below are some result.

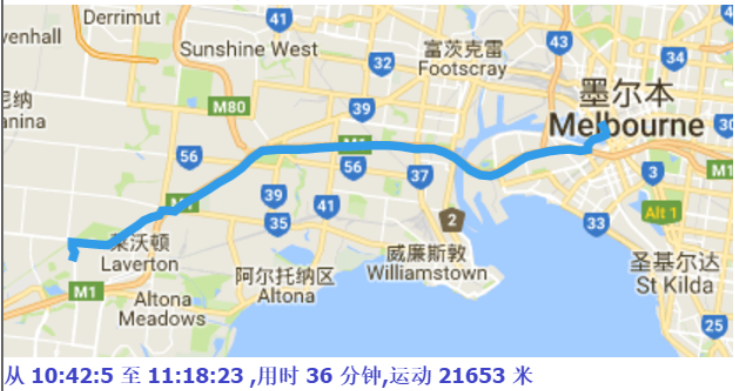
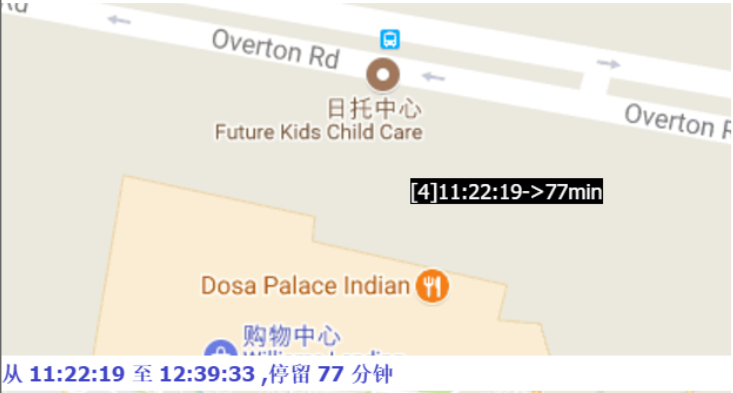
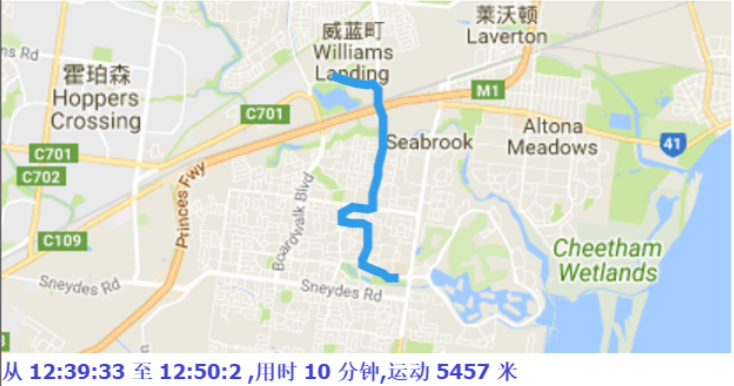
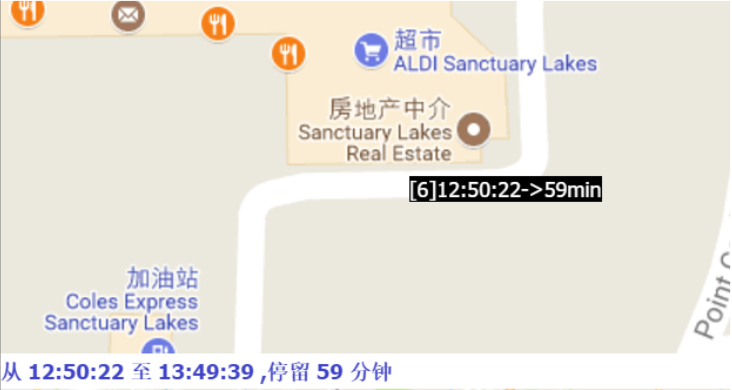

Moreover, if we collect the moving tracjectories of people, It must be a great thing, look at the following picture:
-
trajectory similarity measures
The papers in trajectory similarity measures folder are mainly about how to get the most popular trajectory from a lot of tracjectories. Actually it was very challenging for me to do that from a bunch of dirty data, because every trajectory have different length, different density, different accuracy, and after trying many method, the DTW(dynamic time warping) algorithm seem to work well, so I implement it and belows are some result:


-
interpolation
The papers in interpolation folder are mainly about how to interpolate the latitude and longitude points, traditional interpolation seem only work on one-dimension data, so I implemented and modified piece-wise cubic bessel interpolation in two-dimension, below are the test result:

-
spectral clustering
The papers in spectral clustering folder are mainly about how to aggregate spatial data, when I implemented that algorithm, I found the computation become more and more slowly when the data size growing, further research has realized that it was mainly because of the Jacobi matrix update operation which help to calculate the eigenvector of a matrix. So if someone wants use it in his/her job, parallel the Jacobi update is one way to overcome that problem, below show some result to compare KMeans.
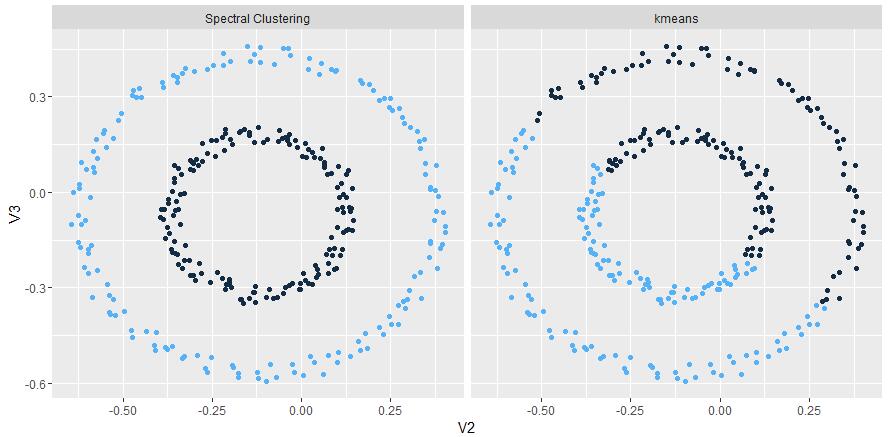
-
HTM(Hierarchical Temporal Memory)
The papers in HTM fouder are mainly about Hierarchical Temporal Memory algorithm which was designed to detect anomaly points from stream data. I useed the nupic framework to implement that algorithm, but as contributors keep submitting code, the project is update very fast, and if someone wants to use it to do something, keep moving with the community. Below show some anomaly detection.
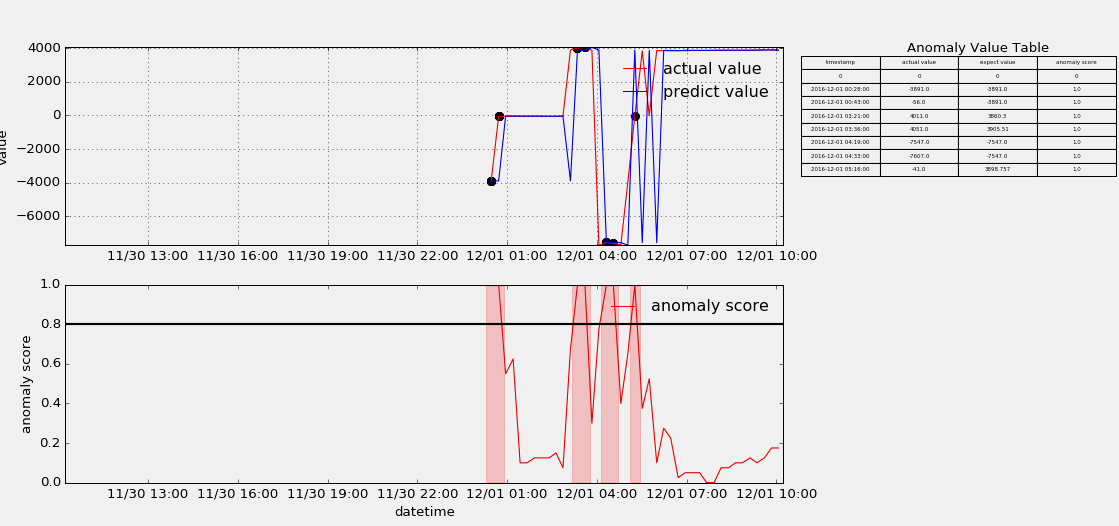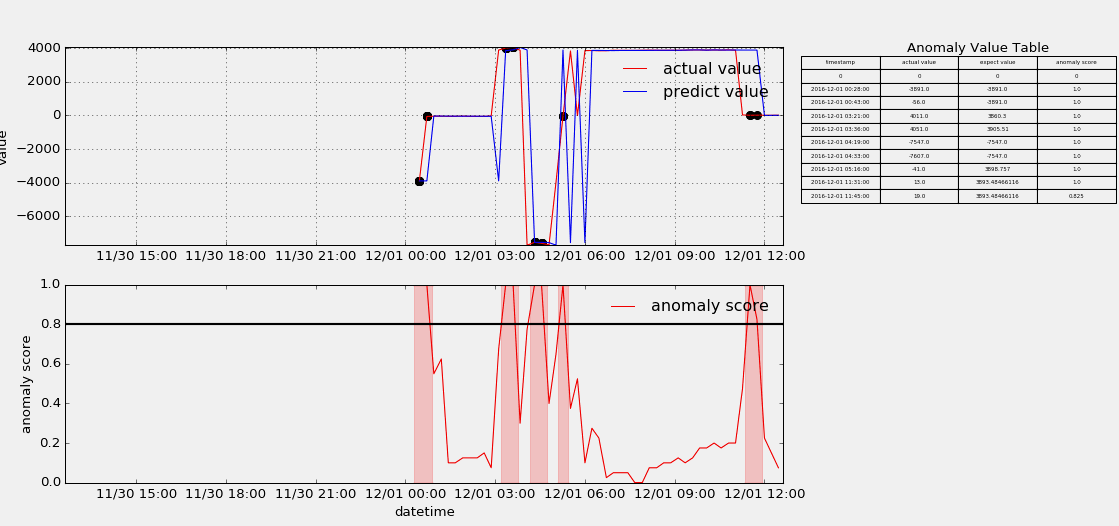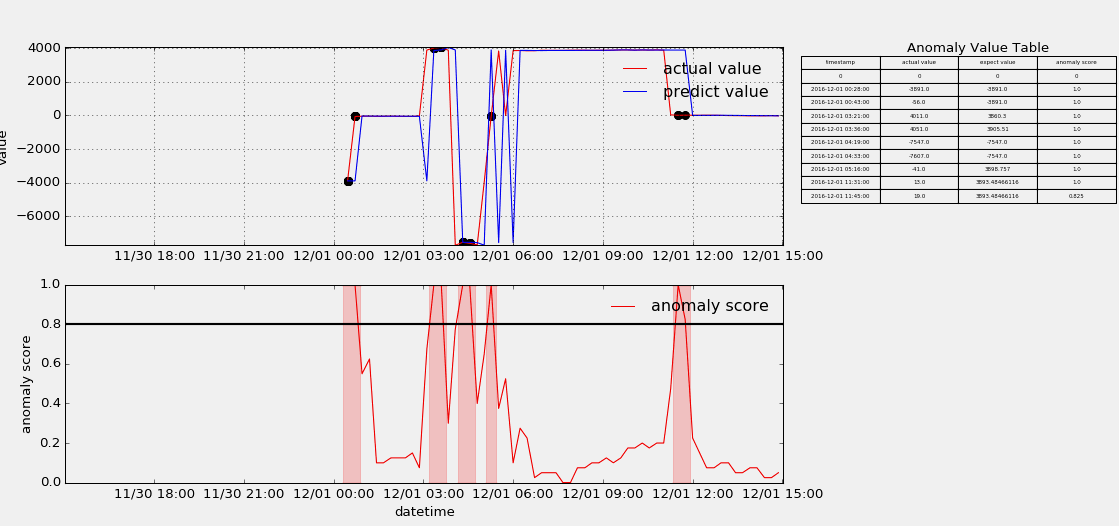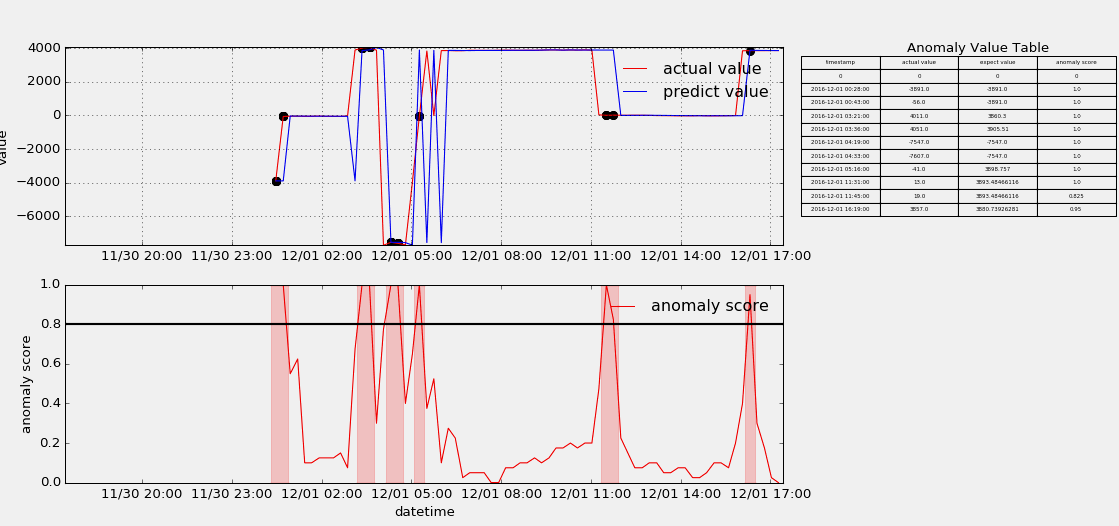
-
CNN(Convolutional Neural Network)
The papers in CNNs fouder are mainly about Convolutional Neural Network included some famous network. I implement some of them in order to distinguish the crossroad pictures. You may curious why we need to do this? Because sometimes it's hard to recognize the crossroad just according to the road data which may contains road type, lng/lat and so on. Below shows some crossroad images, that's what we need to recognize when navigation.



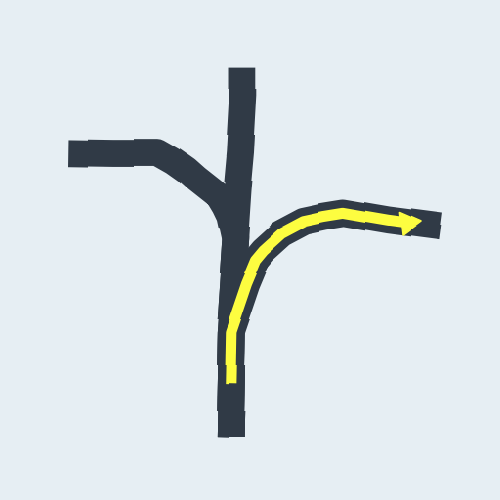



Below shows some non-intersection recognization and key point detection.

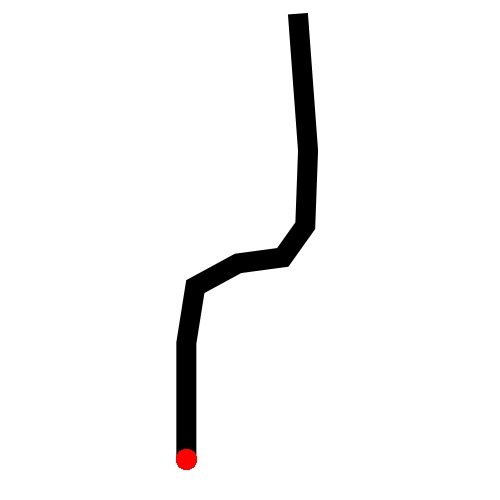




Key point detection.
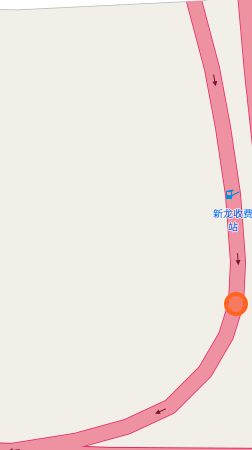
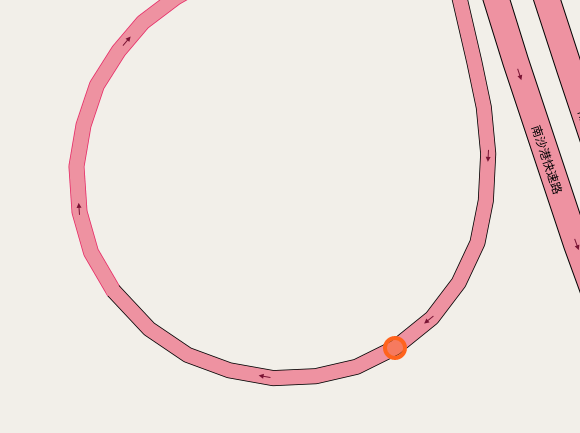
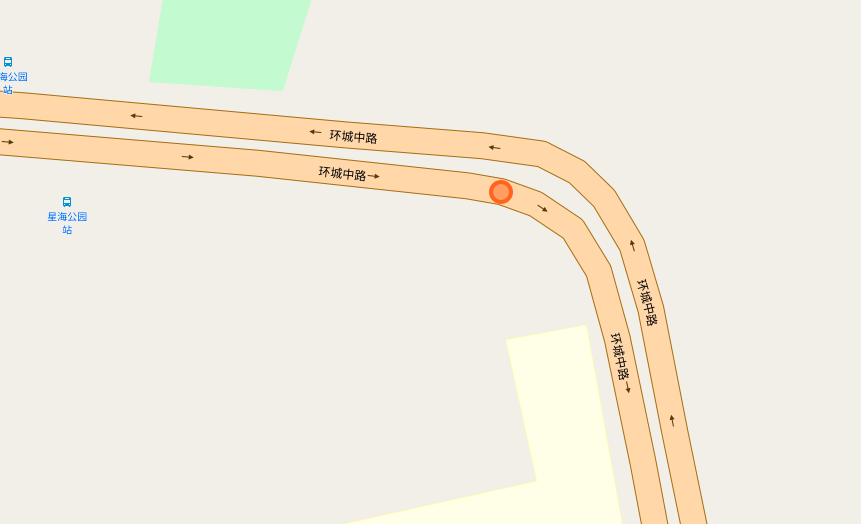
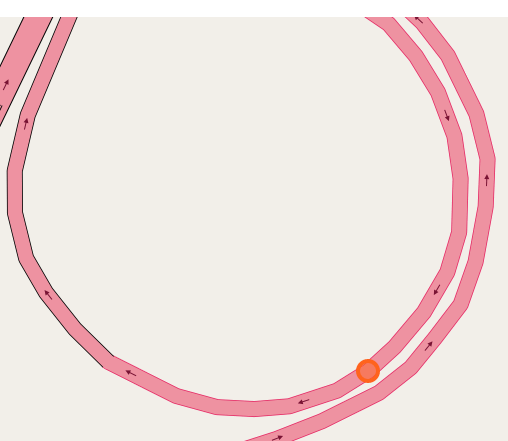

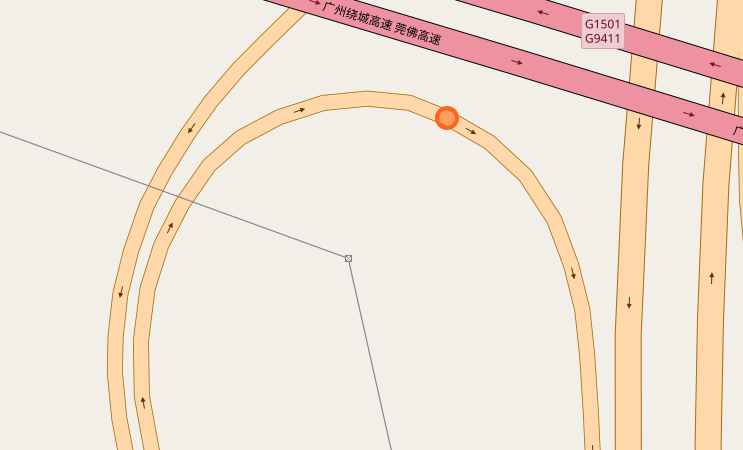
-
RNN(Recurrent Neural Network)
The papers in RNNs are mainly about recurrent neural network, I implement some of them in order to do the recommendation in route planning. After compared GRU,LSTM,BiLSTM and GAN, I found the BiLSTM perform best in generating the recommend route, But it still need more test. Below shows some results generated by the BiLSTM model.
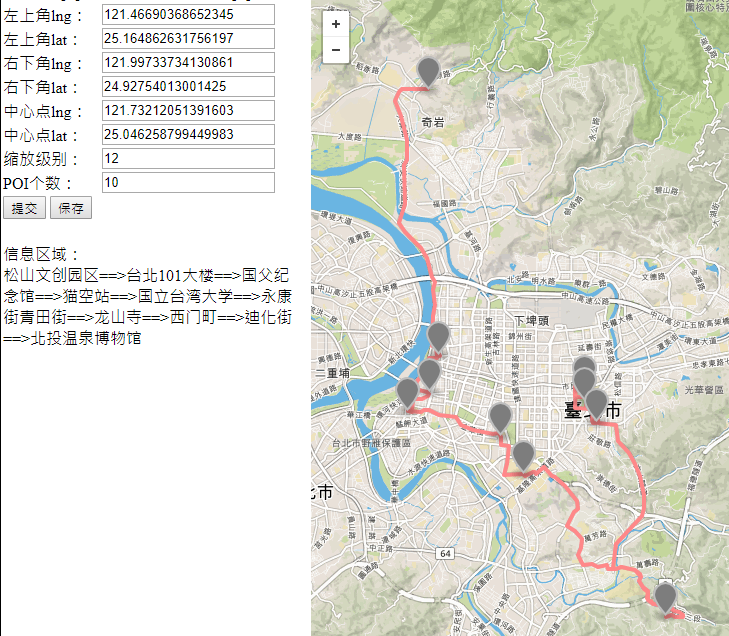
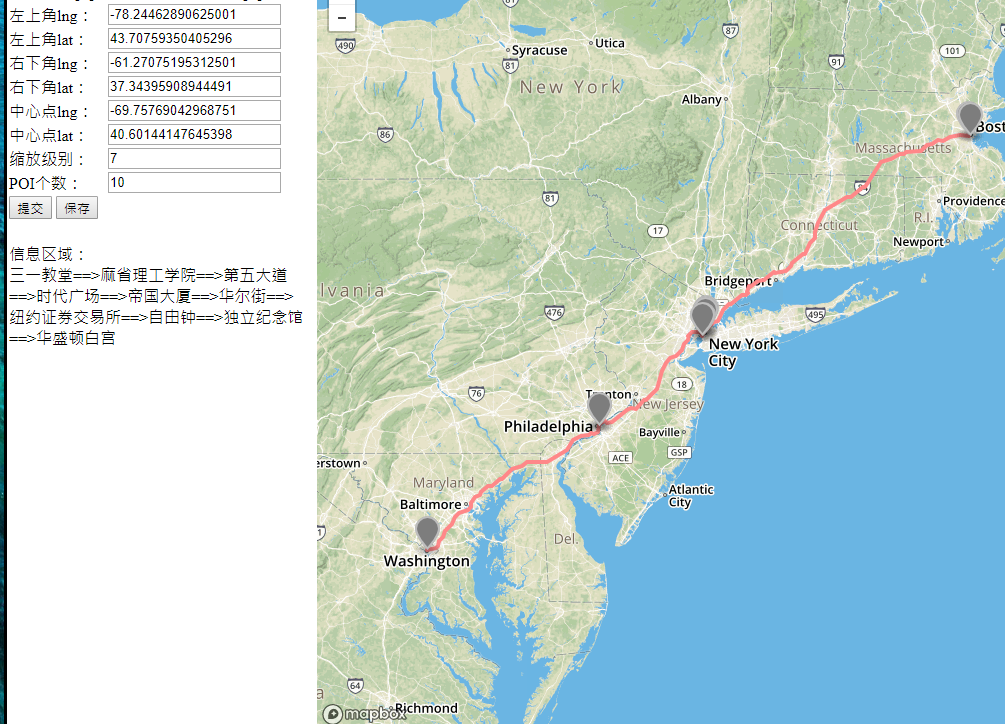
-
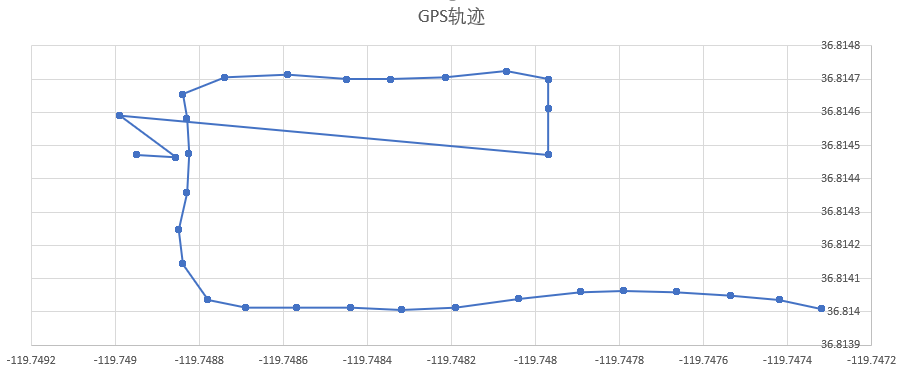
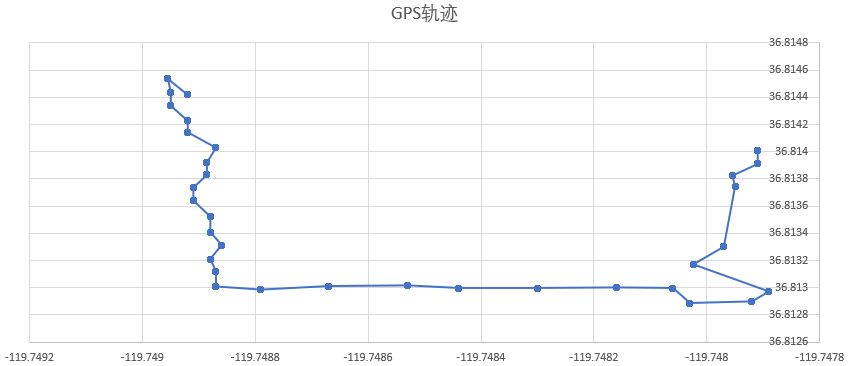
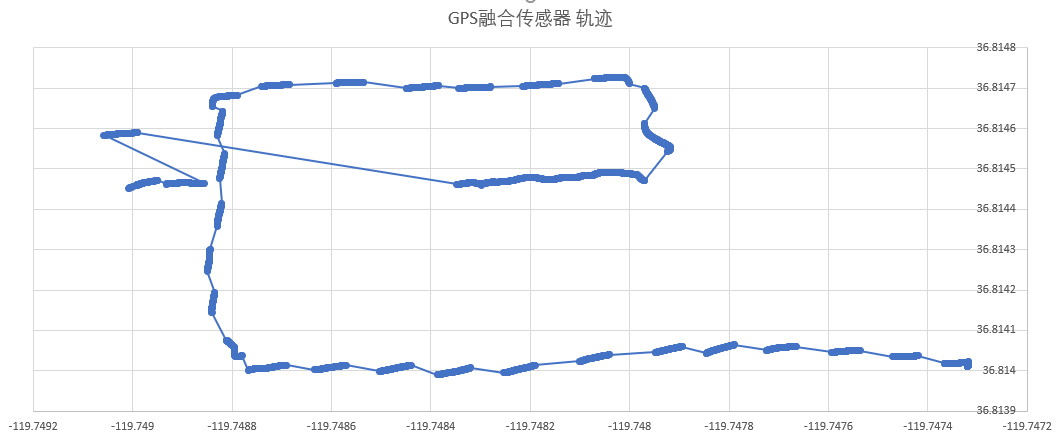
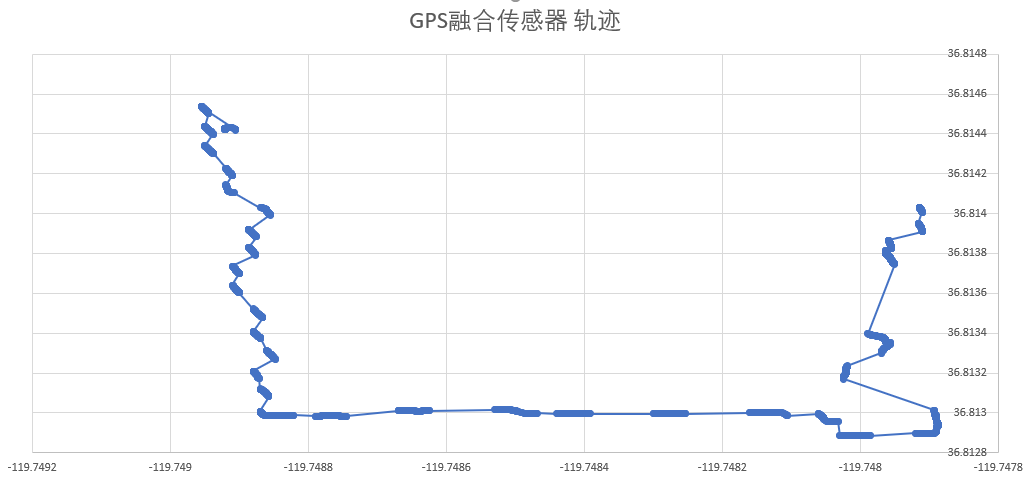
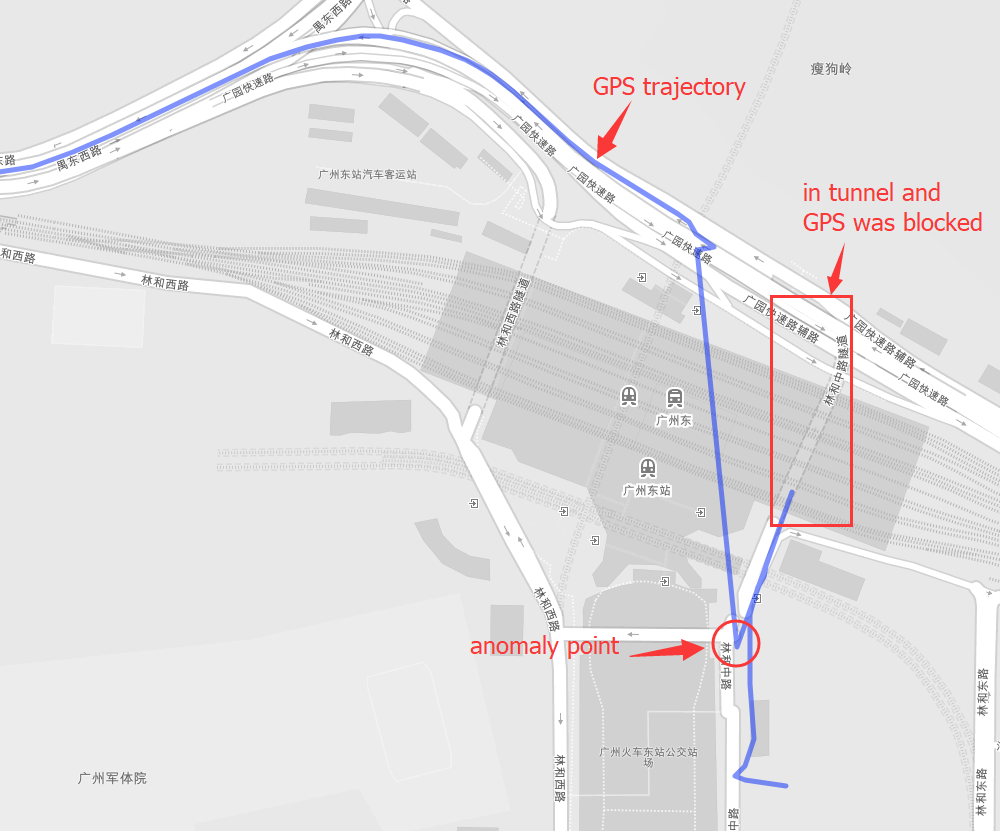
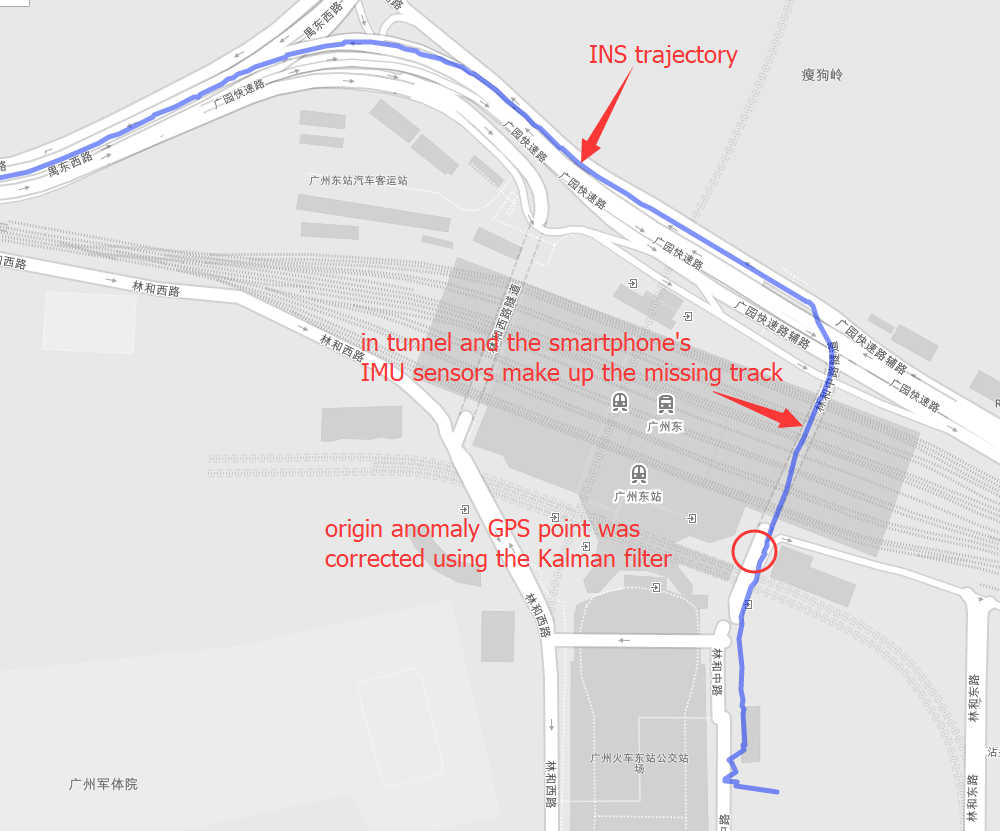
INS(Inertial Navigation System)
The papers in INS are mainly about Inertial Navigation System. Positioning is the most basic step in the driving navigation process, and it is also a very crucial step. An accurate positioning can effectively improve the accuracy of the road-binding, and can also sense the change of the driving pattern more accurately. Since the project is mainly based on mobile phones for navigation and positioning, Currently used is the built-in sensor data (gyroscope, accelerometer, geomagnetic meter, direction sensor, gravity sensor) and GPS data fusion positioning.