CHROMATIN ACTIVATION PROFILING OF STEREOTYPED CHRONIC LYMPHOCYTIC LEUKEMIAS REVEALS A SUBSET #8 SPECIFIC SIGNATURE
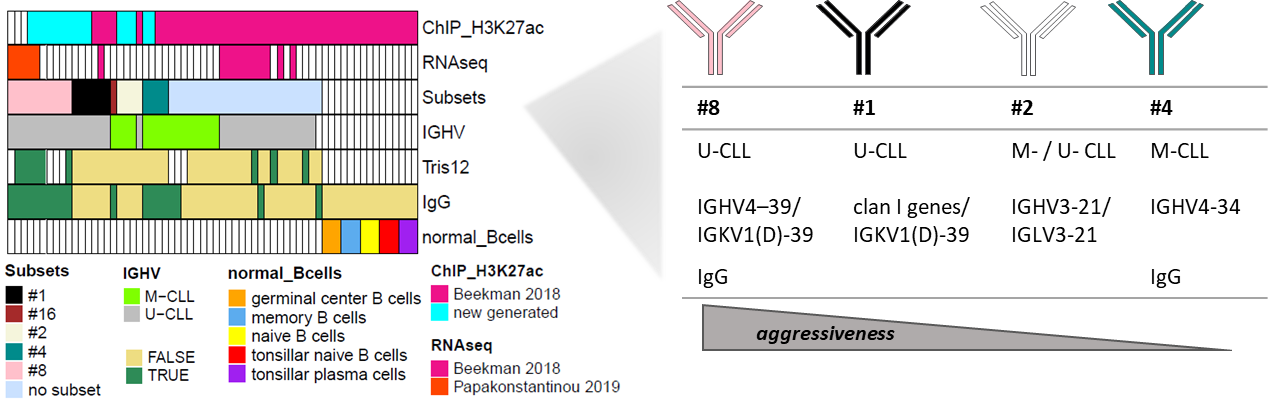
The chromatin activation landscape of major subsets of chronic lymphocytic leukemia (CLL) with stereotyped B-cell receptor immunoglobulin is currently unknown. Here, we report the results of a whole-genome chromatin profiling of histone 3 lysine 27 acetylation of 21 CLLs from subsets #1, #2, #4, and #8 which were compared against non-stereotyped CLLs and normal B cell subpopulations. Although subsets #1, #2, and #4 did not differ much from their non-stereotyped CLL counterparts, subset #8 displayed a remarkably different chromatin activation profile. In particular, we identified 209 de novo active regulatory elements in this subset versus other CLLs or normal B cells, which showed similar patterns with U-CLLs undergoing Richter transformation. These regions were enriched for binding sites of 9 overexpressed transcription factors. In 78/209 regions, we identified 113 candidate overexpressed target genes, being 14% of regions associated with more than two adjacent genes. These included blocks of up to 7 genes, suggesting a local co-upregulation within the same genome compartment. Our findings further underscore the uniqueness of subset #8 CLLs, notable for the highest risk of Richter’s transformation amongst all CLL, and provide additional clues to decipher the molecular basis of its clinical behavior.
The data has been deposited in five levels of organization, from raw to processed data:
- raw data. All the new generated fastq files have been deposited at the European Genome Archive (EGA) under accession id EGAS00001006457
- matrices. All the counts table have been deposited in Zenodo
The packages needed to be installed, in order to run the project are:
install.packages(c("tidyverse", "ggplot2", "stringi", "boot"))
BiocManager::install(c("limma", "ComplexHeatmap", "sva", "DESeq2"))
conda install -c bioconda trim-galore
conda install -c bioconda bwa
conda install -c bioconda picard
conda install -c bioconda samtools
conda install -c bioconda macs2
conda install -c bioconda bedtools
conda install -c bioconda trim-galore
conda install -c bioconda kallisto
input: The resource to find the ChIP seq datapipeline: The pipeline to run the analysis from fastq filesresults: The resource to find the output matrix
input: The resource to find the RNA seq datapipeline: The pipeline to run the analysis from fastq files and the script to import this output into Rresults: The resource to find the output matrix
-
Combat_batchEffectCorrection.R: batch effect correction script -
DiffAnalysis_heatmap_PCA.R: Differential analysis and visualation using complexheatmap and ggplot2 -
Random_resampling.R: random resampling, x100 times differential analysis and reporting the most frequent regions