 Publication: Matcher: An Open-Source Application for Translating Large Structure/Property Data Sets into Insights for Drug Design
Publication: Matcher: An Open-Source Application for Translating Large Structure/Property Data Sets into Insights for Drug Design
Free preprint of above publication
Matcher is a tool for understanding how chemical structure optimization problems have been solved.
Matcher enables deep control over searching structure/activity relationships (SAR) derived from large datasets, and takes the form of an accessible web application with simple deployment.
Matcher is built around the mmpdb platform for matched molecular pair (MMP) analysis. Matcher extends the mmpdb data model, introduces new search algorithms, provides a backend API for executing queries and fetching results, and provides a frontend user interface.
- System Requirements
- Quick Start
- Run Example Query
- Data Included
- Use Different Data
- Metadata Information
- OpenAPI for Backend
- Using mmpdb Commands
- Computer with x86 processor (i.e. most desktop/laptop computers except Apple M-series)
- ARM processors are not yet supported, due to certain dependencies
Clone this repository, and the mmpdb submodule (with git submodule init and git submodule update).
Navigate to the parent matcher directory, then execute:
docker-compose up
Three containers will be launched:
- database container, which contains the PostgreSQL database
- backend container, which is controlled through a FastAPI accessible at localhost:8001
- frontend container, which hosts a Dash app through a Flask API, accessible at localhost:8000
The rate-determining step is for the backend container to use mmpdb commands to process input data, and write resulting MMP data to the database. This process is complete when the below output is observed in the docker-compose console:
INFO:uvicorn.error:Uvicorn running on http://0.0.0.0:8001 (Press CTRL+C to quit)
After the above is observed, in a web browser (Google Chrome or Microsoft Edge), navigate to localhost:8000. The Matcher query interface should load after a few seconds.
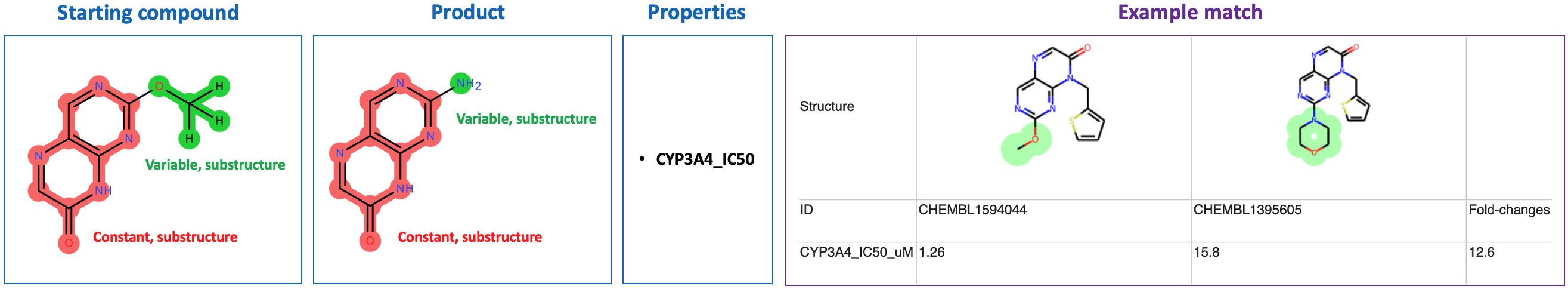
Matcher query logic can be learned by example: click the "Run Example Search" link at the top of the matcher query frontend page (localhost:8000), which will direct to localhost:8000/examples.
Several example inputs/outputs will be displayed. Upon clicking on an example, a new tab will open with the live Matcher interface, containing appropriately-populated input. Output results will load below the input.
Important: The example queries are only guaranteed to work with the example data provided in this package, because these queries specify properties that are present in the example data. If new data is used which has property names differing from the example data's property names, an exception will be thrown in the client js layer, and the example queries will not execute when loaded.
Data is present in the backend/initialize_db directory.
Quick Start data (default): Data filenames begin with "quick_start". Contains 1089 ChEMBL compounds, the minimum to fully reproduce queries described in our publication (TODO: Add hyperlink here).
Rapidly test/debug the deployment: Data filenames begin with "test". Contains 16 ChEMBL compounds, a subset of the Quick Start 1078 compounds, for the purpose of rapid testing during development or troubleshooting. All example queries work, but return only a few results.
Full ChEMBL dataset: Data filenames begin with "ChEMBL_CYP3A4_hERG". Contains 20267 ChEMBL compounds having CYP3A4 inhibition and/or hERG inhibition data, which were included with the mmpdb publication. A superset of the Quick Start data.
The default input compound/property dataset is intentionally very small, so that the containers will initialize quickly for demo purposes.
To use arbitrary data, follow the below steps.
As an example, we illustrate how to use a "medium-size" dataset containing 20,267 compounds taken from the mmpdb publication, and referenced in our publication.
-
Add raw data to the matcher/backend/initialize_db directory. Two files are required, a third file is optional. All files must begin with the same identifier:
your_dataset_name, which in this example isChEMBL_CYP3A4_hERG:- Required: File containing compound SMILES and compound IDs.
- For this example, ChEMBL_CYP3A4_hERG_structures.smi is already included.
- For this example, ChEMBL_CYP3A4_hERG_structures.smi is already included.
- Required: File containing compound IDs and property values.
- For this example, ChEMBL_CYP3A4_hERG_props.txt is already included.
- For this example, ChEMBL_CYP3A4_hERG_props.txt is already included.
- Optional: File containing metadata about the compound property data (whether the data is log transformed, the units, and how the data should be displayed to users).
- For this example, ChEMBL_CYP3A4_hERG_metadata.csv is already included. If you do not wish to provide metadata, edit out the
--metadataargument from this line of code inentrypoint.sh:conda run --no-capture-output -n matcher-api python $MMPDB_DIR/mmpdb.py loadprops -p "${properties}" --metadata "${metadata}" "$postgres_schema\$postgres" && \. If no metadata file is provided, then by default, property labels and data will be displayed to users exactly as provided in the above property value file, and changes between two properties will be treated as differences (B - A).
- For this example, ChEMBL_CYP3A4_hERG_metadata.csv is already included. If you do not wish to provide metadata, edit out the
- Required: File containing compound SMILES and compound IDs.
-
Edit matcher/backend/entrypoint.sh by setting
DATASET=your_dataset_name, usingyour_dataset_namefrom step 1. above.
- Recreate the containers:
- Navigate to the parent directory (matcher/), and execute the following commands (note that using the --volumes flag deletes your previous matcher DB data):
docker-compose down --volumes && \
docker-compose build && \
docker-compose up --force-recreate
This time, around 20 minutes will be required to build the database (depending on computer), due to the larger number of compounds in the input data as compared to the original Quick Start data.
Optionally, property metadata can be passed to the mmpdb loadprops command, for the purpose of customizing how data is displayed to end users in matcher's web UI.
If metadata is not provided, then all data will be displayed as it exists in the database, and all changes will be calculated and displayed as deltas (B-A).
Here is an example property metadata table (values should be tab separated):
property_name base unit display_name display_base display_unit change_displayed
hERG_pIC50 negative_log M hERG_IC50_uM raw uM fold-change
Example:
% mmpdb loadprops --properties hERG.csv --metadata hERG_medatadata.csv 'database$5432$postgres'
In the above case, hERG data is provided in the --properties file as a negative log of molarity, but we provide additional metadata which causes the data to appear to users in the matcher web UI as micromolar IC50 values. This is the most common base/unit conversion use case; not all conversions are supported.
Metadata is stored in columns within the property_name table, and therefore is easy to modify (albeit manually with SQL) even after the database is built.
Supported metadata options are below. All other values, or * value, will be converted to default. None means that the column value will be NULL in property_name table.
base [default=raw]: raw, log, negative_log
unit [default=None]: M, uM
display_name [default=property_name]: characters other than *
display_base [default=raw]: raw
display_unit [default=None]: uM, M
change_displayed [default=delta]: delta, fold-change
Matcher's backend API can be used for querying and gathering results, independently of the frontend, if desired.
The backend API endpoints are documented in backend/openapi.json. This documentation can be viewed when the matcher application is running, at localhost:8001/docs
The matcher database is an extended mmpdb database, and is reverse-compatible with mmpdb commands.
For example, to run mmpdb transform with matcher's database, outputting results to results.csv within your local directory:
First launch matcher as described in Quick Start, then run this command:
docker exec -it \
"$(docker ps | grep 'matcher_backend' | awk '{ print $1 }')" \
conda run -n matcher-api \
python /opt/mmpdb/mmpdb.py transform --smiles 'O=C1NC2=C(C=NC(OC)=N2)N=C1' 'public$postgres' > results.csv