
A simple method for identifying transcription factor mediated regulatory networks from scRNA and scATAC data
- Matthew Moss (Lead)
- Mervin Fansler
- Nicholas Gomez
- Claire Marchal
- Shahin Mohammadi
- Marygrace Trousdell
- Miranda Darby
- Bala Desinghu
- [Samantha Henry]
- Jenelys Ruiz Ortiz
The advent of single cell sequencing technologies have now allowed for the identification and characterization of rare cell types. Identifying the key transcription factors and downstream target genes is important for understanding the biology of these rare populations. The goal of this project is to develop a workflow that identifies and ranks transcriptional regulators important in the various cell states as identified by single cell sequencing. By combining both scRNA-seq and scATAC-seq we can increase our power to identify biologically meaningful gene regulatory networks.
The aim is to take a dataset which includes both single cell RNA and ATAC seqs, identify cell type clusters in them, and then integrate them in order to find different levels of cell type regulators. We would take this opportunity to standardize and automate various aspects of this pipeline, especially the integration between data types, for future uses, and also to allow direct flow into other levels of analysis. Additionally, we hope to provide information to the user about the genes in the identified networks, in order to inform conclusions and future hypothesis/experiment decisions.
The goal of our workflow is to be containerized so that all packages and dependencies are included in the docker image. We only require snakemake to run the pipeline and few R packages in order to visualize the output from MASCARA.
Singularity - a container platform.
Snakemake - a workflow management tool.
R - A software environment for statistical computing and graphics
Install shiny, networkD3, and dplyr within R
install.packages(c("shiny", "networkD3", "dplyr"))
 )
)
A workflow that outputs a table containing a ranked transcription factor mediated network and an easy to use interactive visualization platform of the gene regulatory networks.
Clone this repository using
git clone https://github.com/NCBI-Codeathons/MASCARA.git
The pipeline requires as input:
- scRNA-seq result - a SingleCellExperiment object as .Rds file
- scATAC-seq result - a SingleCellExperiment object as .Rds file
- transcriptome - a GTF file
- chrom.sizes file
- network.tsv - tab-delimited file containing the cluster specific transcription factors and downstream target genes. Column IDs are Celltype, TF (Transcription Factor), TG (Targets), weight (interaction strength on a scale from -1 to 1) , hgnc symble, ensembl gene id, entrez gene id, gene descripton, chromosome, start, stop and strand.

The full example data can be downloaded by navigating to the data/ folder and running
snakemake
This will download two .Rds files, representing the scRNA-seq and scATAC-seq SingleCellExperiment objects from Granja, et al., 2019, as well as GTF and chrom.sizes files for hg19.
Update 11/8/20: Sample data now available for PBMC same cell single cell RNA and ATAC seq which has been verified using Seurat pipelines:
ATAC Meta Data: http://cf.10xgenomics.com/samples/cell-atac/1.0.1/atac_v1_pbmc_10k/atac_v1_pbmc_10k_singlecell.csv
The main pipeline is preconfigured (see config.yaml) to uses these downloaded files. The full pipeline can then be run by navigating to the root of the repository (MASCARA/) and running
snakemake --use-singularity
To run user-supplied data, edit the config.yaml file to specify the locations of the input files (scRNA-seq .Rds, scATAC-seq .Rds, transcriptome GTF, and chrome.sizes). Be sure to also change the genome: value to match the genome that was used in the alignments.
From the root of the repository (MASCARA/), run the pipeline using
snakemake --use-singularity
Once the pipeline has finished running, there will be a final output file data/output/network.tsv. These results can be visualized and explored interactively in a Shiny app by running the following from the command line
Rscript shinyapp/app.R data/output/network.tsv
This will automatically launch open the app in the default web browser.
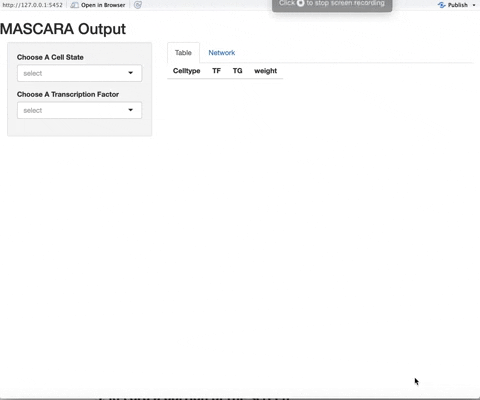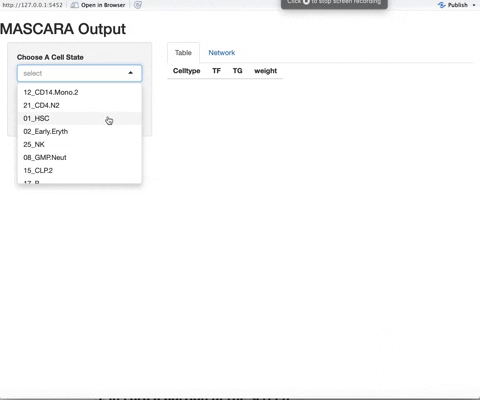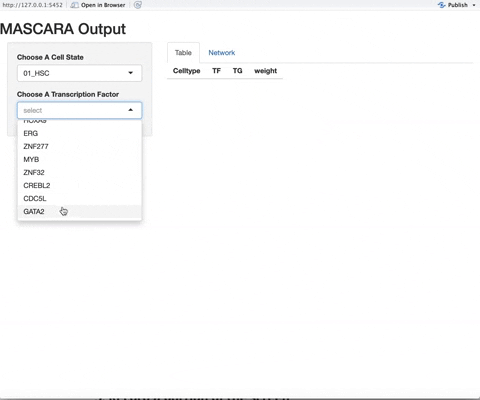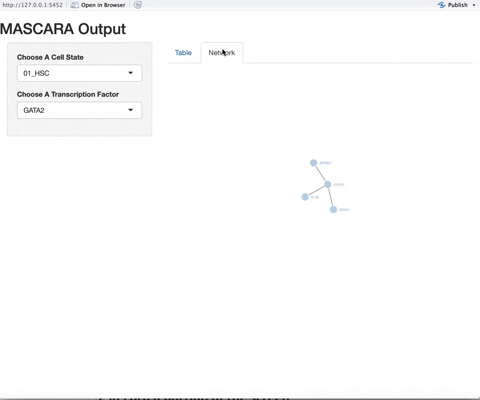
11/8/20 - Added gene information for genes contained within the network, including ensemble gene ID, gene information, and location

In a near future update, we will be adding increased information to the network visualization, including hover over information for the genes and motif/tissue specificity information for the transcription factors.
Longer term goals include to integrate a pseudotime analysis as a method to understand how regulatory networks change at different time points during cell type differentiation and or disease progression. Incorporating a trajectory inference may help to better characterize the evolution and divergences between cell clusters.
Data used in tutorial:
- Granja, J.M., Klemm, S., McGinnis, L.M. et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat Biotechnol 37, 1458–1465 (2019) doi:10.1038/s41587-019-0332-7
Packages/Applications
- Docker - Dirk Merkel. 2014. Docker: lightweight Linux containers for consistent development and deployment. Linux J. 2014, 239, Article 2 (March 2014).
- Cicero - Pliner, H. A., Packer, J. S., McFaline-Figueroa, J. L., Cusanovich, D. A., Daza, R. M., Aghamirzaie, D., … Trapnell, C. (2018). Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Molecular cell, 71(5), 858–871.e8. doi:10.1016/j.molcel.2018.06.044
- ChromVar - Schep, A., Wu, B., Buenrostro, J. et al. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978 (2017) doi:10.1038/nmeth.4401
- ACTIONet - Mohammadi, S., Davila-Velderrain, J., Kellis, M. (2019) A multiresolution framework to characterize single-cell state landscapes. bioRxiv 746339; doi: doi.org/10.1101/746339
- Biomart - Durinck S, Spellman P, Birney E, Huber W (2009). “Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt.” Nature Protocols, 4, 1184–1191.
