{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
List of participants and affiliations:
- Daniel Paiva Agustinho, Baylor College of Medicine (Team Leader)
- Sushant Potdar, University of Arkansas (Writer)
- Alexis Quintero, National University of Colombia
- Andrew Miller, University of Utah
- John Arnn, Utah Public Health Laboratory
- Rong Guo, University of Maryland, College Park
- Participant, Affiliation
- Project Goals
- Introduction
- Approach
- Results
- Future Work
This project aims to identify the common and unique SARS-CoV-2 variants in clinical isolates and wastewater samples from a particular geographical area for a specific time period. The aim is to understand if wastewater samples can predict variants found in clinical isolates or vice-versa. A detailed introduction for the project is below.
The analysis of the genomic sequence of clinical isolates of SARS-CoV-2 can provide insights into circulating strains in the human population. However, this approach may present certain limitations. Clinical isolates primarily represent severe cases that required hospitalization, potentially leading to a biased view of the virus's diversity. As a result, the genomic sequences obtained from clinical isolates may not fully capture the prevalence and variation of milder or asymptomatic infections, which constitute the majority of cases in the population.
In contrast, wastewater sequencing offers a more inclusive approach, providing a broader representation of circulating strains without bias towards severe cases. Wastewater contains genetic material shed by individuals with varying degrees of infection, including asymptomatic and mild cases, making it more reflective of the overall viral diversity in a community. However, wastewater testing has its challenges. The conditions within wastewater can lead to RNA degradation, which may result in the potential underestimation of minor alleles or variants. The indiscriminate representation of genetic material in wastewater also includes viral RNA from non-infectious particles, making it necessary to carefully interpret the results.
In this project, we aim to compare wastewater sampling with clinical isolates from specific temporal and geographical subsets. Our goal is to evaluate the representativeness of each approach concerning the total population of variants found circulating in these subsets. By assessing the genomic sequences obtained from both wastewater and clinical isolates, we can gain valuable insights into the diversity and prevalence of SARS-CoV-2 strains within each community. This comparative analysis will help us understand the strengths and limitations of each approach, making it possible to provide a more comprehensive view of the viral dynamics in distinct populations. To start with, we first extracted data from New York, USA for the year 2022, and then we repeated the same approach for various geographical locations and created a summary table for year 2022. We also estimated ratios of the number of variants only represent in wastewater among the states of Florida, New York, Texas and Wisconsin in year 2022.
As a test case, we first parsed the COVID-19 VCF file from AWS for samples from New York in the year 2022 using the query_sql.txt code and extracted the biosamples, accession numbers, chromosomes (chr), position (pos), reference sequence (ref), alternative sequence (alt), read depth (dp), effect (the type of variation), codon (nucleotide sequence change), and g_ad_2 (alt allele depth). For more info on these different attributes (and more), you can check this link: https://www.ncbi.nlm.nih.gov/sra/docs/sra-cloud-based-annotated-variations-table/
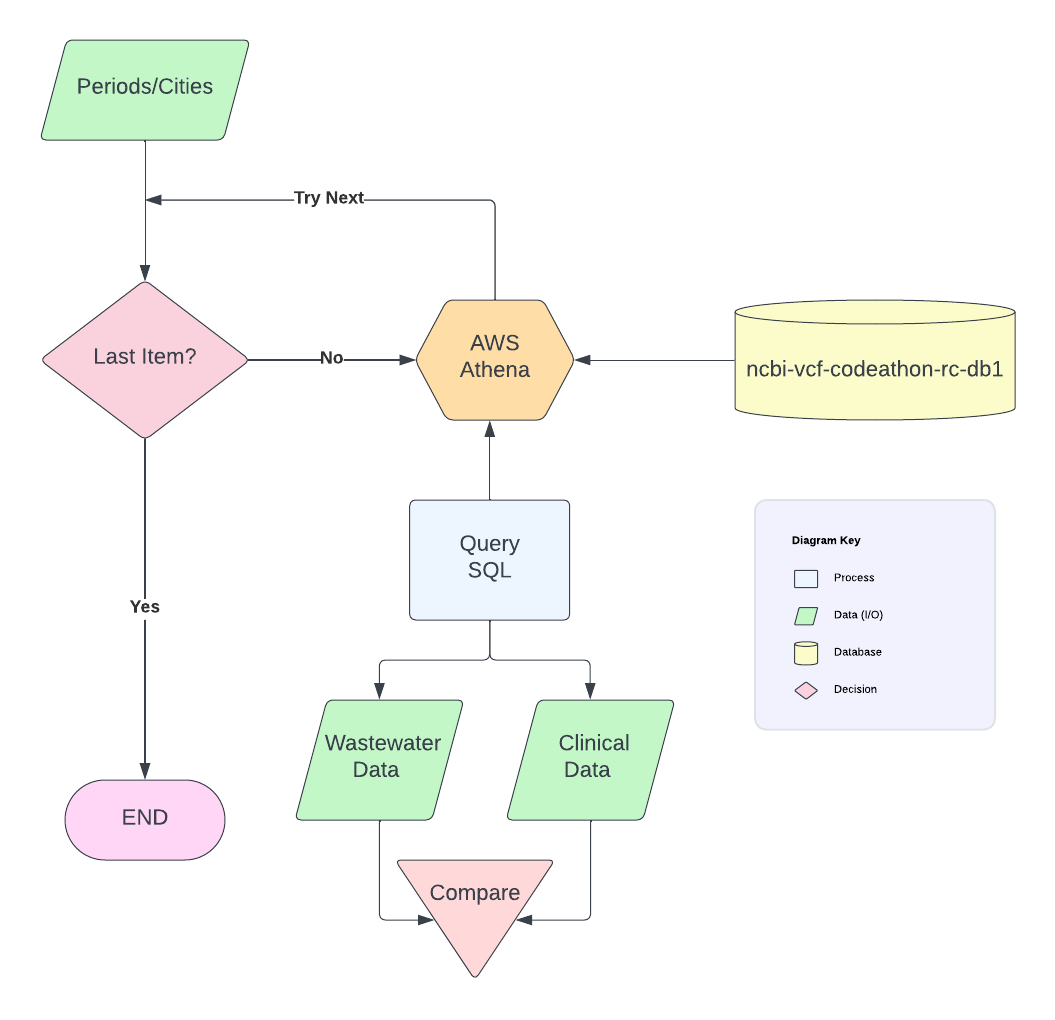
We later used a customized R script (ci_ww_variants.R) and removed any duplicated rows within the clinical isolates or the wastewater biosamples. We then identified the common variants present between wastewater and clinical isolates, as well as the unique variants present in each of these samples.
We repeated this for various geographical locations for the year 2022, and created a CovidSummary table with all the months of the year separated for the different geographical locations. Some filters were applied to create a CovidSummary_filter table: synonymous and intergenic variants were removed; allele frequency cut-off >= 0.4 to filter out low frequency alleles that were caused by sequencing errors or other technical errors.
In addition, we estimated a ratio = # of variants only in wastewater/ all # of variants in wastewater to evaluate the representativeness of the clinical samples. The higher the number means more variants only represent in the wastewater, but were not captured in the clinical samples.
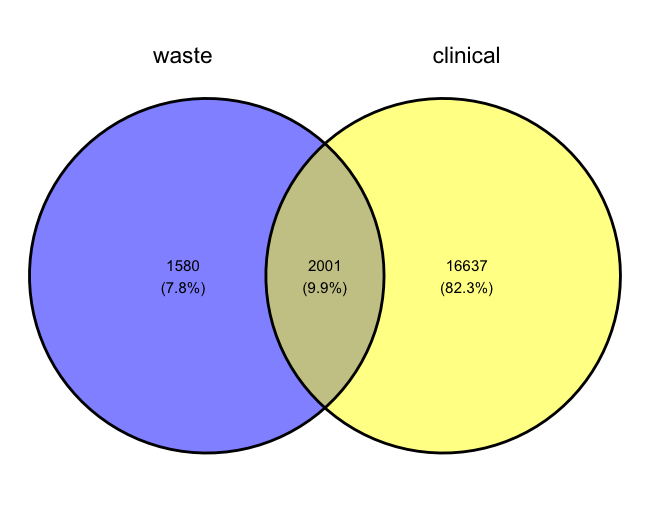
We found that around 82% of total variants from our dataset were unique to the clinical isolates, 8% of total variants were unique to the wastewater samples. 10% of the variants were common between the two sample types in New York for the year 2022.
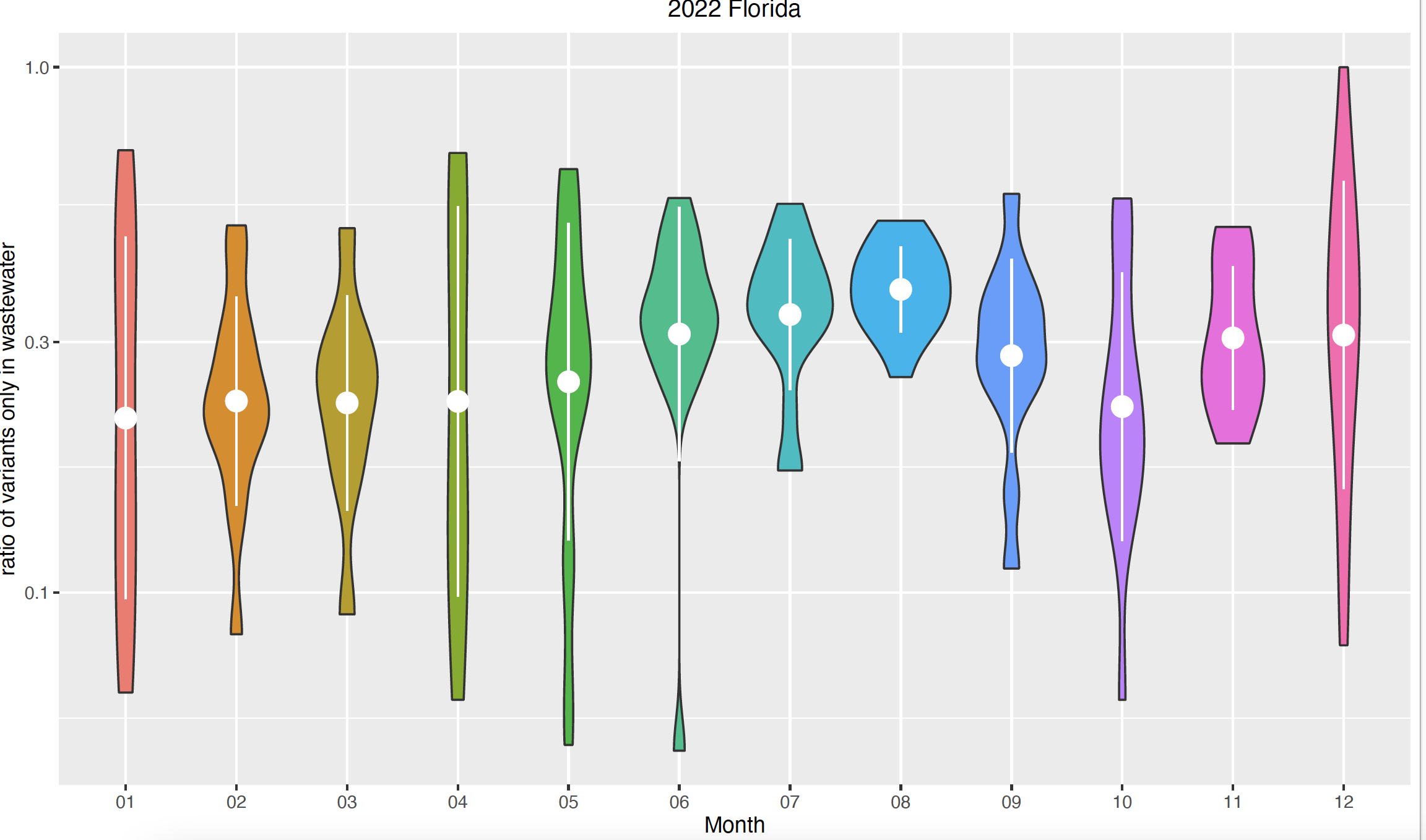
As an example, when we look at the distribution of the ratio of variants that are only present in Florida's wastewater across the 12 months in 2022 (using unfiltered data), we find that during the summer months, the ratio is high with the highest confidence, compared to other months in the year.
For more information and results, please see the 'finalpresentation.pdf' file in our main repository.