

![]()
FastANI is developed for fast alignment-free computation of whole-genome Average Nucleotide Identity (ANI). ANI is defined as mean nucleotide identity of orthologous gene pairs shared between two microbial genomes. FastANI supports pairwise comparison of both complete and draft genome assemblies. Its underlying procedure follows a similar workflow as described by Goris et al. 2007. However, it avoids expensive sequence alignments and uses Mashmap as its MinHash based sequence mapping engine to compute the orthologous mappings and alignment identity estimates. Based on our experiments with complete and draft genomes, its accuracy is on par with BLAST-based ANI solver and it achieves two to three orders of magnitude speedup. Therefore, it is useful for pairwise ANI computation of large number of genome pairs. More details about its speed, accuracy and potential applications are described here: "High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries".
Clone the software from Github and follow INSTALL.txt to compile the code. There is also an option to download dependency-free binary for Linux or OSX through the latest release.
- Produce help page. Quickly check the software usage and available command line options.
$ ./fastANI -h- One to One. Compute ANI between single query and single reference genome:
$ ./fastANI -q [QUERY_GENOME] -r [REFERENCE_GENOME] -o [OUTPUT_FILE] Here QUERY_GENOME and REFERENCE_GENOME are the query genome assemblies in fasta or multi-fasta format.
- One to Many. Compute ANI between single query genome and multiple reference genomes:
$ ./fastANI -q [QUERY_GENOME] --rl [REFERENCE_LIST] -o [OUTPUT_FILE]For above use case, REFERENCE_LIST should be a file containing directory paths to reference genomes, one per line.
- Many to Many. When there are multiple query genomes and multiple reference genomes:
$ ./fastANI --ql [QUERY_LIST] --rl [REFERENCE_LIST] -o [OUTPUT_FILE]Again, QUERY_LIST and REFERENCE_LIST are files containing paths to genomes, one per line.
Output format. In all above use cases, OUTPUT_FILE will contain tab delimited row(s) with query genome, reference genome, ANI value, count of bidirectional fragment mappings, and total query fragments. Alignment fraction (wrt. the query genome) is simply the ratio of mappings and total fragments. Optionally, users can also get a second .matrix file with identity values arranged in a phylip-formatted lower triangular matrix by supplying --matrix parameter. NOTE: No ANI output is reported for a genome pair if ANI value is much below 80%. Such case should be computed at amino acid level.
Two genome assemblies are provided in data folder to do a quick test run.
We suggest users to do an adequate quality check of their input genome assemblies (both reference and query), especially the N50 be ≥10 Kbp.
- One to One. Here we compute ANI between Escherichia coli and Shigella flexneri genomes provided in the data folder.
$ ./fastANI -q data/Shigella_flexneri_2a_01.fna -r data/Escherichia_coli_str_K12_MG1655.fna -o fastani.out Expect output log in the following format in the console:
$ ./fastANI -q data/Shigella_flexneri_2a_01.fna -r data/Escherichia_coli_str_K12_MG1655.fna -o fastani.out
>>>>>>>>>>>>>>>>>>
Reference = [data/Escherichia_coli_str_K12_MG1655.fna]
Query = [data/Shigella_flexneri_2a_01.fna]
Kmer size = 16
Fragment length = 3000
Threads = 1
ANI output file = fastani.out
>>>>>>>>>>>>>>>>>>
....
....
INFO, skch::main, Time spent post mapping : 0.00310319 secOutput is saved in file fastani.out, provided above using the -o option.
$ cat fastani.out
data/Shigella_flexneri_2a_01.fna data/Escherichia_coli_str_K12_MG1655.fna 97.7507 1303 1608Above output implies that the ANI estimate between S. flexneri and E. coli genomes is 97.7507. Out of the total 1608 sequence fragments from S. flexneri genome, 1303 were aligned as orthologous matches.
FastANI supports visualization of the reciprocal mappings computed between two genomes.
Getting this visualization requires a one to one comparison using FastANI as discussed above, except an additional flag --visualize should be provided.
This flag forces FastANI to output a mapping file (with .visual extension) that contains information of all the reciprocal mappings.
Finally, an R script is provided in the repository which uses genoPlotR package to plot these mappings.
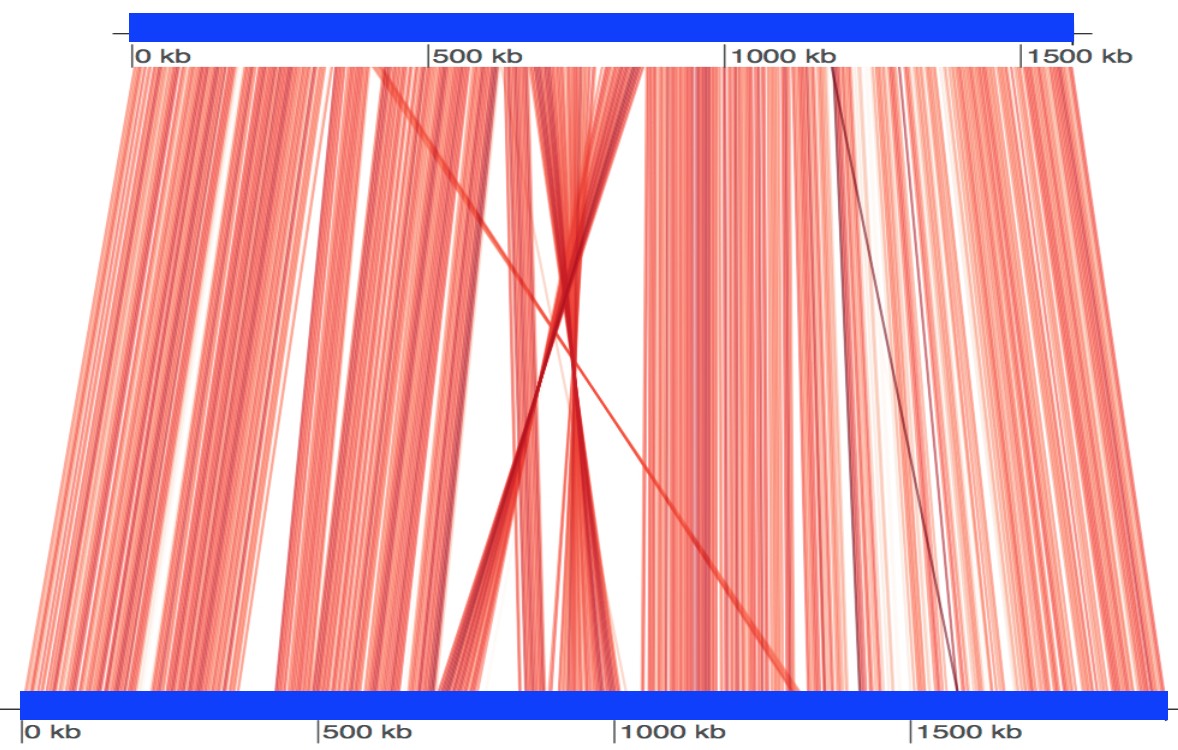
Here we show an example run using two genomes: Bartonella quintana (GenBank: CP003784.1) and Bartonella henselae (NCBI Reference Sequence: NC_005956.1).
$ ./fastANI -q B_quintana.fna -r B_henselae.fna --visualize -o fastani.out
$ Rscript scripts/visualize.R B_quintana.fna B_henselae.fna fastani.out.visualUsing above commands, we get a plot file fastani.out.visual.pdf displayed below. Each red line segment denotes a reciprocal mapping between two genomes, indicating their evolutionary conserved regions. Also see this page.
FastANI (v1.1 onwards) supports multi-threading, see the help page on how to configure thread count. To parallelize FastANI beyond single compute node, users also have the choice to simply divide their reference database into multiple chunks, and execute them as parallel processes. We provide a script in the repository to randomly split the database for this purpose.
Users are welcome to report any issue or feedback related to FastANI by posting a Github issue.