Home
The Trinity Cancer Transcriptome Analysis Toolkit (CTAT) includes a suite of tools focused on identifying and characterizing fusion transcripts in cancer. The tools and comprehensive workflow for fusion study via Trinity CTAT is illustrated below:
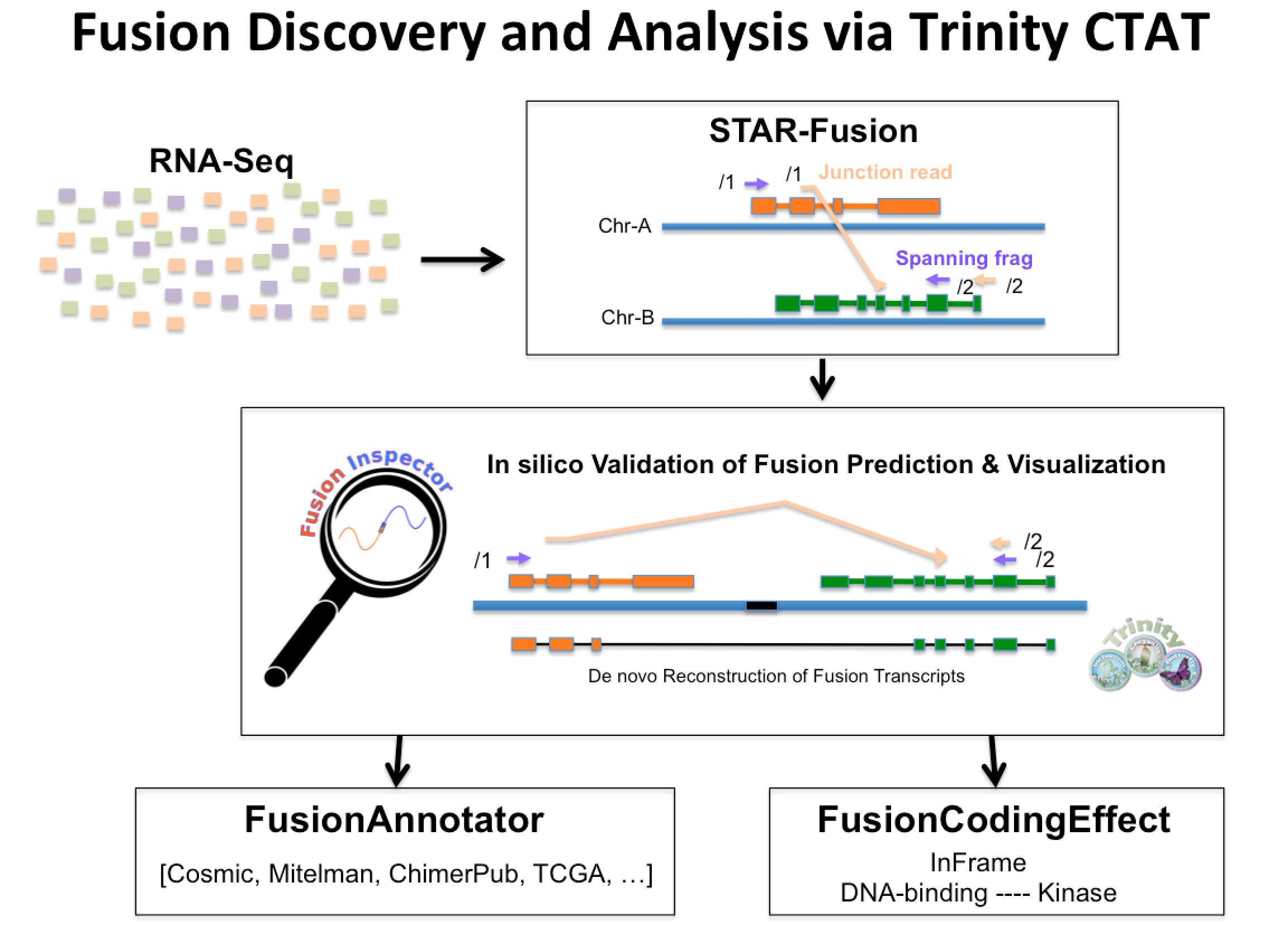
Tools leveraged within CTAT include STAR-Fusion, FusionInspector, and Trinity. The tutorial below demonstrates the use of each of these utilities in identifying and characterizing cancer fusion transcripts. Note that the STAR-Fusion software incorporates FusionInspector and several companion utilities that will be used for fusion analysis.
The Trinity CTAT Fusion workflow involves first running STAR-Fusion to identify candidate fusion transcripts based on discordant read alignments. Predicted fusions are then 'in silico validated' using FusionInspector, which performs a more refined exploration of the candidate fusion transcripts, runs Trinity to de novo assemble fusion transcripts from the RNA-Seq reads, and provides the evidence in a suitable format to facilitate visualization. Predicted fusions are annotated according to prior knowledge of fusion transcripts relevant to cancer biology (or previously observed in normal samples and less likely to be relevant to cancer), and assessed for the impact of the predicted fusion event on coding regions, indicating whether the fusion is in-frame or frame-shifted along with combinations of domains that are expected to exist in the chimeric protein.
The tutorial includes a small data set that can be leveraged using modest computational resources, so it should run on a any hardware having at least 4G of RAM (so suitable for most contemporary personal computing machines). The tutorial data consists of the following files:
Genome data:
minigenome.fa : small genome sequence consisting of ~750 genes.
minigenome.gtf : transcript structure annotations for these genes.
RNA-Seq data:
rnaseq_1.fastq.gz : RNA-Seq read 1 data ('left' read)
rnaseq_2.fastq.gz : RNA-Seq read 2 data ('right' read)
Meta data:
CTAT_HumanFusionLib.mini.dat.gz : a small fusion annotation library
Obtain the tutorial data by downloading it from the tutorial release page, or you can obtain it using 'git' like so:
# Optionally, obtain data set using git:
% git clone https://github.com/STAR-Fusion/STAR-Fusion-Tutorial.git
To run the full tutorial, you would need the following software installed (unless you use Docker (see below)):
Note, each tool above may have it's own additional software installation dependencies, such as samtools, jellyfish, salmon, etc... It's easiest to just use our Docker image, if that's an option, as everything's already installed in it.
Our trinityctat/ctatfusion Docker image contains all the above tools and their dependencies. If you have Docker installed, you can use it very effectively here.
# First, cd into the STAR-Fusion-Tutorial directory (or whatever it's named in your download)
# Then, run Docker like so:
% docker run --rm -it -v `pwd`:/data -v /tmp:/tmp trinityctat/starfusion:latest bash
# The above should put you into the Docker environment.
# Then go to the /data directory, where you'll find the tutorial data
% cd /data/STAR-Fusion-Tutorial
Using the Trinity CTAT ecosystem of tools requires a CTAT genome lib, which is effectively a resource package containing a target genome, reference annotations, and various meta data files that support fusion-finding. Normally (outside of this tutorial context), you would use a pre-compiled CTAT genome lib provided to you (as indicated in the STAR-Fusion documentation). If you have a different target than the entire human genome, as is the case here, then we would need to build a custom CTAT fusion lib.
To build our custom tutorial-supporting CTAT genome lib, run the following along with using the provided tutorial data files:
Below, we refer to ${STAR_FUSION_HOME} as the path to the base installation directory for STAR-Fusion. In the Docker image, this is set as an environmental variable, so you can refer to it directly as $STAR_FUSION_HOME. If you're not using Docker, consider setting this as an environmental variable for convenience.
% ${STAR_FUSION_HOME}/ctat-genome-lib-builder/prep_genome_lib.pl \
--genome_fa minigenome.fa \
--gtf minigenome.gtf \
--fusion_annot_lib CTAT_HumanFusionLib.mini.dat.gz \
--dfam_db human \
--pfam_db current
# takes ~45 minutes
Running the above will create a 'ctat_genome_lib_build_dir/' directory and populate it with the reference data, perform blast searches to identify sequence similar sequences, and build some databases that will be leveraged by CTAT fusion tools.
IMPORTANT: This ctat genome lib should only be used with this tutorial. It is not complete and will not allow for good performance with regular data sets. While it takes an hour to build the above abridged genome lib, it can take a much longer time (a day or more) for building the comprehensive genome lib - which is why we suggest using the downloadable plug-n-play version for standard use where possible.
Run STAR-Fusion to predict fusions like so:
% ${STAR_FUSION_HOME}/STAR-Fusion \
--left_fq rnaseq_1.fastq.gz \
--right_fq rnaseq_2.fastq.gz \
--genome_lib_dir ctat_genome_lib_build_dir
# takes a couple minutes
By default, the outputs are written to a subdirectory 'STAR-Fusion_outdir', where you'll find the two primary output files:
star-fusion.fusion_predictions.tsv # fusion predictions including the identity of all evidence reads
star-fusion.fusion_predictions.abridged.tsv # shortened version of the above, lacking the voluminous read identities
Take a look at the format of the abridged output file:
% head STAR-Fusion_outdir/star-fusion.fusion_predictions.abridged.tsv
#FusionName JunctionReadCount SpanningFragCount SpliceType LeftGene LeftBreakpoint RightGene RightBreakpoint LargeAnchorSupport FFPM LeftBreakDinuc LeftBreakEntropy RightBreakDinuc RightBreakEntropy annots
TATDN1--GSDMB 81 126 ONLY_REF_SPLICE TATDN1^TATDN1 minigenome:6584842:- GSDMB^GSDMB minigenome:2142920:- YES_LDAS 282.2894 GT 1.9219 AG 1.5628 ["CCLE","Klijn_CellLines","FA_CancerSupp","ChimerPub","INTRACHROMOSOMAL[minigenome:4.40Mb]"]
TATDN1--GSDMB 30 126 ONLY_REF_SPLICE TATDN1^TATDN1 minigenome:6584842:- GSDMB^GSDMB minigenome:2138981:- YES_LDAS 212.7398 GT 1.9219 AG 1.9086 ["CCLE","Klijn_CellLines","FA_CancerSupp","ChimerPub","INTRACHROMOSOMAL[minigenome:4.40Mb]"]
ACACA--STAC2 31 42 ONLY_REF_SPLICE ACACA^ACACA minigenome:1869270:- STAC2^STAC2 minigenome:2067875:- YES_LDAS 99.5513 GT 1.9656 AG 1.9656 ["ChimerSeq","CCLE","Klijn_CellLines","FA_CancerSupp","INTRACHROMOSOMAL[minigenome:0.07Mb]","LOCAL_REARRANGEMENT:-:[69422]"]
CCDC6--RET 28 14 ONLY_REF_SPLICE CCDC6^CCDC6 minigenome:609384:- RET^RET minigenome:490475:+ YES_LDAS 57.2761 GT 1.8892 AG 1.8323 ["ChimerSeq","ChimerKB","FA_CancerSupp","Cosmic","Mitelman","ChimerPub","HaasMedCancer","Larsson_TCGA","YOSHIHARA_TCGA","INTRACHROMOSOMAL[minigenome:0.08Mb]","LOCAL_INVERSION:-:+:[82786]"]
BCAS4--BCAS3 8 84 ONLY_REF_SPLICE BCAS4^BCAS4 minigenome:4187310:+ BCAS3^BCAS3 minigenome:2490128:+ NO_LDAS 125.4619 GT 1.6402 AG 1.9899 ["ChimerPub","ChimerSeq","chimerdb_pubmed","CCLE","FA_CancerSupp","INTRACHROMOSOMAL[minigenome:1.68Mb]"]
BCAS4--BCAS3 4 84 ONLY_REF_SPLICE BCAS4^BCAS4 minigenome:4187310:+ BCAS3^BCAS3 minigenome:2485565:+ NO_LDAS 120.0071 GT 1.6402 AG 1.3996 ["ChimerPub","ChimerSeq","chimerdb_pubmed","CCLE","FA_CancerSupp","INTRACHROMOSOMAL[minigenome:1.68Mb]"]
RPS6KB1--SNF8 18 21 ONLY_REF_SPLICE RPS6KB1^RPS6KB1 minigenome:2354713:+ SNF8^SNF8 minigenome:2265737:- YES_LDAS 53.185 GT 1.3753 AG 1.8323 ["Klijn_CellLines","FA_CancerSupp","ChimerSeq","CCLE","INTRACHROMOSOMAL[minigenome:0.09Mb]","LOCAL_INVERSION:+:-:[87595]"]
RP11-208G20.2--PSPHP1 12 42 INCL_NON_REF_SPLICE RP11-208G20.2^RP11-208G20.2 minigenome:6394049:+ PSPHP1^PSPHP1 minigenome:6324196:+ YES_LDAS 73.6407 AA 1.3996 TA 1.8323 INTRACHROMOSOMAL[minigenome:0.07Mb],LOCAL_REARRANGEMENT:+:[69653]
TOB1--SYNRG 17 22 ONLY_REF_SPLICE TOB1^TOB1 minigenome:2296883:- SYNRG^SYNRG minigenome:1999668:- YES_LDAS 53.185 GT 1.4566 AG 1.8892 ["FA_CancerSupp","CCLE","INTRACHROMOSOMAL[minigenome:0.24Mb]"]
The format of the fusion output is described in the STAR-Fusion documentation. The first few columns identify the fused genes and the number of reads supporting the fusion prediction as split-reads or spanning fragments.
FusionInspector performs in silico validation of fusion transcripts by realigning reads to both the whole genome and to fusion contigs where fusion candidate genes are positioned adjacent to one another in the order of their putative fusion event. Fusion reads that would align discordantly in the whole genome should instead align concordantly to the fusion contigs. FusionInspector identifies such reads and re-scores the fusion predictions. The fusion contigs along with the aligned reads are a useful product for visualizing and inspecting the evidence supporting the fusions.
FusionInspector comes bundled with the STAR-Fusion software and can be launched via the STAR-Fusion interface. FusionInspector has two modes: 'inspect' or 'validate' mode. In 'inspect' mode, it just aligns the STAR-Fusion identified evidence reads to the fusion contigs for inspection purposes. In 'validate' mode, it aligns all reads simultaneously to the whole genome and to the fusion contigs to identify those reads that align concordantly as fusion evidence to the fusion contigs.
Run FusionInspector via the STAR-Fusion interface like so:
% ${STAR_FUSION_HOME}/STAR-Fusion \
--left_fq rnaseq_1.fastq.gz \
--right_fq rnaseq_2.fastq.gz \
--genome_lib_dir ctat_genome_lib_build_dir \
--FusionInspector validate
# takes ~10 minutes
All FusionInspector outputs will be found in the directory 'STAR-Fusion_outdir/FusionInspector-validate/'
Examine the files found there:
% ls -ltr STAR-Fusion_outdir/FusionInspector-validate/
The most relevant output files include:
finspector.fusion_predictions.final # the 'in silico validated' fusion predictions
finspector.fusion_predictions.final.abridged.FFPM.annotated # the abridged version including annotations
and files that are useful for viewing in IGV:
finspector.bed # reference transcript structure annotations in BED format
finspector.consolidated.bam # reads aligned to the fusion contigs
finspector.junction_reads.bam # junction / split-reads supporting fusions
finspector.spanning_reads.bam # fusion spanning fragment evidence
Load these files into IGV for inspecting the evidence supporting the fusions.
If you need the IGV software, you can launch it from here - just find the 'Launch' button in the center of that linked web page.
This FusionInspector view of the data would look like so:

More details about FusionInspector can be found on the FusionInspector documentation wiki.
Use Trinity to de novo reconstruct fusion transcripts like so:
% ${STAR_FUSION_HOME}/STAR-Fusion \
--left_fq rnaseq_1.fastq.gz \
--right_fq rnaseq_2.fastq.gz \
--genome_lib_dir ctat_genome_lib_build_dir \
--FusionInspector validate \
--denovo_reconstruct
# takes ~5 minutes
Now, reexamine the contents of the 'STAR-Fusion_outdir/FusionInspector-validate/' directory:
% ls -ltr STAR-Fusion_outdir/FusionInspector-validate/
You'll find additional outputs:
finspector.gmap_trinity_GG.fusions.fasta # de novo reconstructed fusion transcripts
finspector.gmap_trinity_GG.fusions.gff3.bed # fusion transcript structure in the fusion contig context
The 'finspector.gmap_trinity_GG.fusions.gff3.bed' can be uploaded into IGV for viewing along with the read evidence.
To explore the effect the fusion events have on coding regions of the fused genes, you can run:
% ${STAR_FUSION_HOME}/STAR-Fusion \
--left_fq rnaseq_1.fastq.gz \
--right_fq rnaseq_2.fastq.gz \
--genome_lib_dir ctat_genome_lib_build_dir \
--examine_coding_effect
# takes a second
You should now find an output file 'STAR-Fusion_outdir/star-fusion.fusion_predictions.abridged.coding_effect.tsv' that includes additional columns describing the impact of the fusion event on the coding regions. Below, the extra columns and some examples are shown, with the fields transposed for easy viewing:
0 #FusionName
1 JunctionReadCount
2 SpanningFragCount
3 SpliceType
4 LeftGene
5 LeftBreakpoint
6 RightGene
7 RightBreakpoint
8 LargeAnchorSupport
9 FFPM
10 LeftBreakDinuc
11 LeftBreakEntropy
12 RightBreakDinuc
13 RightBreakEntropy
14 annots
15 CDS_LEFT_ID
16 CDS_LEFT_RANGE
17 CDS_RIGHT_ID
18 CDS_RIGHT_RANGE
19 PROT_FUSION_TYPE
20 FUSION_MODEL
21 FUSION_CDS
22 FUSION_TRANSL
23 PFAM_LEFT
24 PFAM_RIGHT
0 ARFGEF2--SULF2
1 2
2 13
3 ONLY_REF_SPLICE
4 ARFGEF2^ARFGEF2
5 minigenome:4071002:+
6 SULF2^SULF2
7 minigenome:4059796:-
8 NO_LDAS
9 20.4557
10 GT
11 1.6895
12 AG
13 1.9656
14 ["Klijn_CellLines","FA_CancerSupp","CCLE","INTRACHROMOSOMAL[minigenome:0.00Mb]","LOCAL_INVERSION:+:-:[3001]"]
15 ARFGEF2^ENST00000371917.4
16 1-121
17 SULF2^ENST00000359930.4
18 176-2610
19 INFRAME
20 minigenome|+|[0]4070882-4071002[0]<==>minigenome|-|[2]4020222-4020249[2]|[1]4021256-4021309[2]|[1]4021554-4021587[1]|[0]4023630-4023753[0]|[2]4024927-4025069[1]|[0]4025310-4025479[2]|[1]4025987-4026046[2]|[1]4027056-4027150[0]|[2]4027714-4027810[2]|[1]4028117-4028345[1]|[0]4031209-4031404[0]|[2]4035505-4035634[2]|[1]4036089-4036145[2]|[1]4037687-4037815[2]|[1]4042005-4042180[0]|[2]4043442-4043592[2]|[1]4049137-4049306[0]|[2]4051324-4051475[1]|[0]4059557-4059796[1]
21 atgcaggagagccagaccaagagcatgttcgtgtcccgggccctggagaagatcctagccgacaaggaggtgaagcggccccagcactcccagctgcgcagggcctgccaggtggcgctcgGTTCCATGCAGGTGATGAACAAGACCCGGCGCATCATGGAGCAGGGCGGGGCGCACTTCATCAACGCCTTCGTGACCACACCCATGTGCTGCCCCTCACGCTCCTCCATCCTCACTGGCAAGTACGTCCACAACCACAACACCTACACCAACAATGAGAACTGCTCCTCGCCCTCCTGGCAGGCACAGCACGAGAGCCGCACCTTTGCCGTGTACCTCAATAGCACTGGCTACCGGACAGCTTTCTTCGGGAAGTATCTTAATGAATACAACGGCTCCTACGTGCCACCCGGCTGGAAGGAGTGGGTCGGACTCCTTAAAAACTCCCGCTTTTATAACTACACGCTGTGTCGGAACGGGGTGAAAGAGAAGCACGGCTCCGACTACTCCAAGGATTACCTCACAGACCTCATCACCAATGACAGCGTGAGCTTCTTCCGCACGTCCAAGAAGATGTACCCGCACAGGCCAGTCCTCATGGTCATCAGCCATGCAGCCCCCCACGGCCCTGAGGATTCAGCCCCACAATATTCACGCCTCTTCCCAAACGCATCTCAGCACATCACGCCGAGCTACAACTACGCGCCCAACCCGGACAAACACTGGATCATGCGCTACACGGGGCCCATGAAGCCCATCCACATGGAATTCACCAACATGCTCCAGCGGAAGCGCTTGCAGACCCTCATGTCGGTGGACGACTCCATGGAGACGATTTACAACATGCTGGTTGAGACGGGCGAGCTGGACAACACGTACATCGTATACACCGCCGACCACGGTTACCACATCGGCCAGTTTGGCCTGGTGAAAGGGAAATCCATGCCATATGAGTTTGACATCAGGGTCCCGTTCTACGTGAGGGGCCCCAACGTGGAAGCCGGCTGTCTGAATCCCCACATCGTCCTCAACATTGACCTGGCCCCCACCATCCTGGACATTGCAGGCCTGGACATACCTGCGGATATGGACGGGAAATCCATCCTCAAGCTGCTGGACACGGAGCGGCCGGTGAATCGGTTTCACTTGAAAAAGAAGATGAGGGTCTGGCGGGACTCCTTCTTGGTGGAGAGAGGCAAGCTGCTACACAAGAGAGACAATGACAAGGTGGACGCCCAGGAGGAGAACTTTCTGCCCAAGTACCAGCGTGTGAAGGACCTGTGTCAGCGTGCTGAGTACCAGACGGCGTGTGAGCAGCTGGGACAGAAGTGGCAGTGTGTGGAGGACGCCACGGGGAAGCTGAAGCTGCATAAGTGCAAGGGCCCCATGCGGCTGGGCGGCAGCAGAGCCCTCTCCAACCTCGTGCCCAAGTACTACGGGCAGGGCAGCGAGGCCTGCACCTGTGACAGCGGGGACTACAAGCTCAGCCTGGCCGGACGCCGGAAAAAACTCTTCAAGAAGAAGTACAAGGCCAGCTATGTCCGCAGTCGCTCCATCCGCTCAGTGGCCATCGAGGTGGACGGCAGGGTGTACCACGTAGGCCTGGGTGATGCCGCCCAGCCCCGAAACCTCACCAAGCGGCACTGGCCAGGGGCCCCTGAGGACCAAGATGACAAGGATGGTGGGGACTTCAGTGGCACTGGAGGCCTTCCCGACTACTCAGCCGCCAACCCCATTAAAGTGACACATCGGTGCTACATCCTAGAGAACGACACAGTCCAGTGTGACCTGGACCTGTACAAGTCCCTGCAGGCCTGGAAAGACCACAAGCTGCACATCGACCACGAGATTGAAACCCTGCAGAACAAAATTAAGAACCTGAGGGAAGTCCGAGGTCACCTGAAGAAAAAGCGGCCAGAAGAATGTGACTGTCACAAAATCAGCTACCACACCCAGCACAAAGGCCGCCTCAAGCACAGAGGCTCCAGTCTGCATCCTTTCAGGAAGGGCCTGCAAGAGAAGGACAAGGTGTGGCTGTTGCGGGAGCAGAAGCGCAAGAAGAAACTCCGCAAGCTGCTCAAGCGCCTGCAGAACAACGACACGTGCAGCATGCCAGGCCTCACGTGCTTCACCCACGACAACCAGCACTGGCAGACGGCGCCTTTCTGGACACTGGGGCCTTTCTGTGCCTGCACCAGCGCCAACAATAACACGTACTGGTGCATGAGGACCATCAATGAGACTCACAATTTCCTCTTCTGTGAATTTGCAACTGGCTTCCTAGAGTACTTTGATCTCAACACAGACCCCTACCAGCTGATGAATGCAGTGAACACACTGGACAGGGATGTCCTCAACCAGCTACACGTACAGCTCATGGAGCTGAGGAGCTGCAAGGGTTACAAGCAGTGTAACCCCCGGACTCGAAACATGGACCTGGGACTTAAAGATGGAGGAAGCTATGAGCAATACAGGCAGTTTCAGCGTCGAAAGTGGCCAGAAATGAAGAGACCTTCTTCCAAATCACTGGGACAACTGTGGGAAGGCTGGGAAGGT
22 MQESQTKSMFVSRALEKILADKEVKRPQHSQLRRACQVALGSMQVMNKTRRIMEQGGAHFINAFVTTPMCCPSRSSILTGKYVHNHNTYTNNENCSSPSWQAQHESRTFAVYLNSTGYRTAFFGKYLNEYNGSYVPPGWKEWVGLLKNSRFYNYTLCRNGVKEKHGSDYSKDYLTDLITNDSVSFFRTSKKMYPHRPVLMVISHAAPHGPEDSAPQYSRLFPNASQHITPSYNYAPNPDKHWIMRYTGPMKPIHMEFTNMLQRKRLQTLMSVDDSMETIYNMLVETGELDNTYIVYTADHGYHIGQFGLVKGKSMPYEFDIRVPFYVRGPNVEAGCLNPHIVLNIDLAPTILDIAGLDIPADMDGKSILKLLDTERPVNRFHLKKKMRVWRDSFLVERGKLLHKRDNDKVDAQEENFLPKYQRVKDLCQRAEYQTACEQLGQKWQCVEDATGKLKLHKCKGPMRLGGSRALSNLVPKYYGQGSEACTCDSGDYKLSLAGRRKKLFKKKYKASYVRSRSIRSVAIEVDGRVYHVGLGDAAQPRNLTKRHWPGAPEDQDDKDGGDFSGTGGLPDYSAANPIKVTHRCYILENDTVQCDLDLYKSLQAWKDHKLHIDHEIETLQNKIKNLREVRGHLKKKRPEECDCHKISYHTQHKGRLKHRGSSLHPFRKGLQEKDKVWLLREQKRKKKLRKLLKRLQNNDTCSMPGLTCFTHDNQHWQTAPFWTLGPFCACTSANNNTYWCMRTINETHNFLFCEFATGFLEYFDLNTDPYQLMNAVNTLDRDVLNQLHVQLMELRSCKGYKQCNPRTRNMDLGLKDGGSYEQYRQFQRRKWPEMKRPSSKSLGQLWEGWEG
23 DCB-PARTIAL|23-121~|1.8e-41^DUF1981|69-95|0.37^HEAT|73-95|0.013
24 Sulfatase-PARTIAL|~176-374|3.5e-52^Phosphodiest-PARTIAL|~176-320|0.00054^Mannosyl_trans2|187-211|1.1^DUF4976|377-395|0.023^DUF5082|420-444|0.5^DUF3740|528-664|2.7e-52^CCDC73|619-667|8.5e-05^DUF5082|631-654|0.001^DUF3740|693-709|5.6^Mannosyl_trans2|745-793|0.00023^DUF4976|777-815|0.0014
No... you can actually have one command that includes all relevant parameters and you can run STAR-Fusion followed by FusionInspector, de novo reconstruction, and explore coding region impacts like so:
% ${STAR_FUSION_HOME}/STAR-Fusion \
--left_fq rnaseq_1.fastq.gz \
--right_fq rnaseq_2.fastq.gz \
--genome_lib_dir ctat_genome_lib_build_dir \
--FusionInspector validate \
--denovo_reconstruct \
--examine_coding_effect
Congratulations on making it through the CTAT Fusion tutorial. For more information on Trinity CTAT and to stay current with our activities and developments, visit our Trinity CTAT page and join our Google Forum