TransDecoder missing obvious ORFs? #114
Comments
|
Hi,
The issue is because of the -m 1 parameter. I should probably make it so
that it can't be run with -m less than 50 to avoid this issue.
TransDecoder doesn't predict short peptides.... the scoring system isn't
suited to it. If it's 50 aa or greater, should work fine.
best,
~b
…On Mon, Sep 28, 2020 at 6:13 AM Lior Glick ***@***.***> wrote:
I am running TransDecoder 5.5.0 (installed using conda) to predict plant
proteins from transcripts.
I used the commands:
TransDecoder.LongOrfs -t transcripts.fasta -m 1
TransDecoder.Predict -t transcripts.fasta
I notice some strange behavior. For example, one of the records in
transcripts.fasta is:
>transcript:AT1G13607.1.mrna1
ATGAACAATTTTCGAATAACCATTGTTGCGTTCTTAGCCGTTCTTGTCTTCACCACAACT
GTTACGAATTCTTTGGATGAACCTAATATGGACACTATATCCAAATCCAGAGAATACAAA
TGTAAAATTGACCTGGATTGTTCAAACCACATTGCATGTAGGCATTGTTCTTATCGCAAT
TGCAAATGCGATCATGGAACCTGTAAATGCATGCCATGACTCTTAACCCACAAGGCCACA
AGCCATTGATCCAACTGCATCTTCAACTCGTCTTAATCTCTCCTATATATGTACTCTTTT
GTTTGTAATGCAAAAGAAAATAAAACATAATATTTTCAGTTGATAAACTACTAATGAAAT
ATTATACGTCAACGAAATTTAGTATATAAACTACAAAACGGCAAAAATAGCTTTCTCGAA
ACCAACAAAGTTAATTGGACAAACGACAAAAA
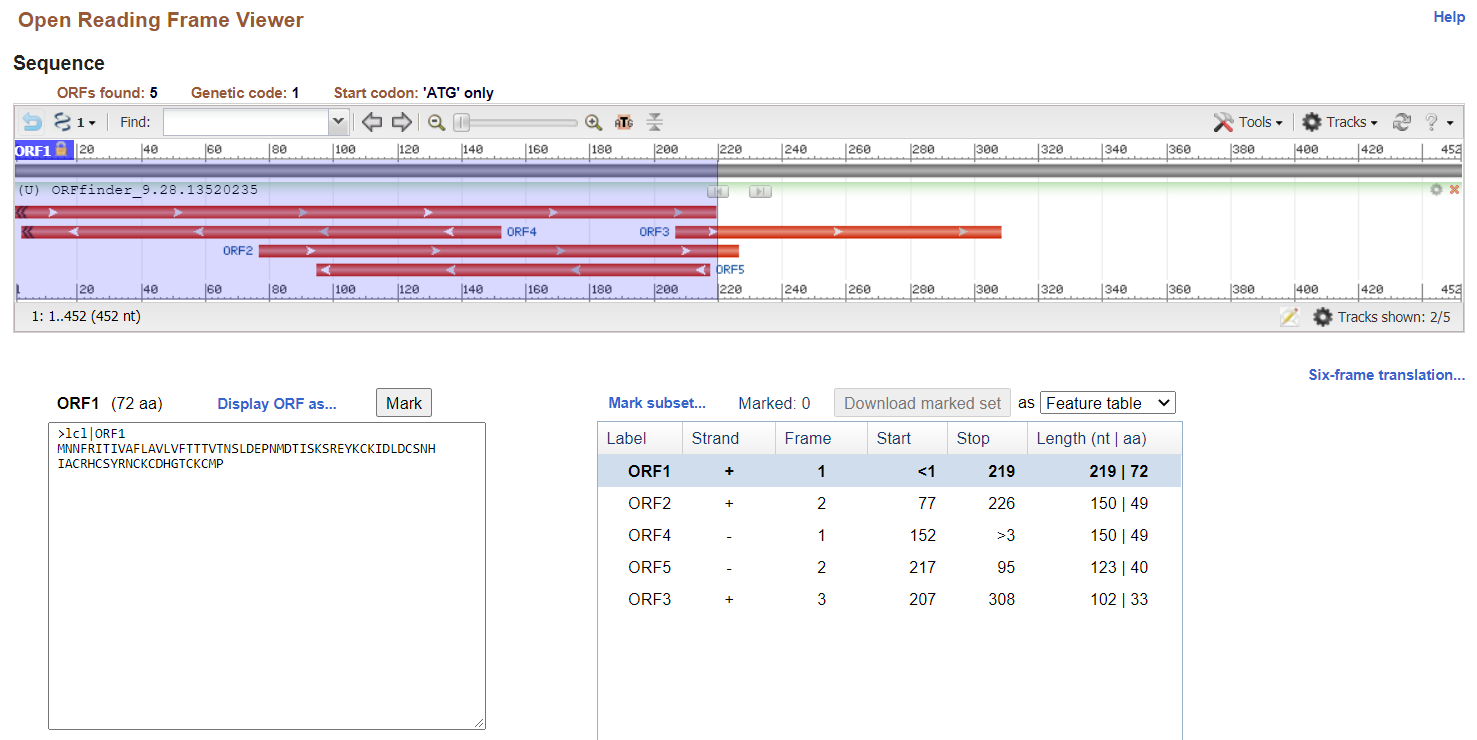
When I run this sequence through NCBI's ORF finder
<https://www.ncbi.nlm.nih.gov/orffinder/>, I get several peptides,
including a 72 AA long one which I assume is correct.
[image: image]
<https://user-images.githubusercontent.com/5146503/94420047-433c3300-018c-11eb-8e7a-9edc262404c1.png>
However, relevant predictions from TransDecoder's output look like this:
>transcript:AT1G13607.1.mrna1.p10 GENE.transcript:AT1G13607.1.mrna1~~transcript:AT1G13607.1.mrna1.p10 ORF type:5prime_partial len:20 (-),score=0.90 transcript:AT1G13607.1.mrna1:391-450(-)
FVVCPINFVGFEKAIFAVL*
>transcript:AT1G13607.1.mrna1.p18 GENE.transcript:AT1G13607.1.mrna1~~transcript:AT1G13607.1.mrna1.p18 ORF type:complete len:2 (+),score=0.25 transcript:AT1G13607.1.mrna1:120-125(+)
M*
>transcript:AT1G13607.1.mrna1.p8 GENE.transcript:AT1G13607.1.mrna1~~transcript:AT1G13607.1.mrna1.p8 ORF type:complete len:23 (-),score=4.50 transcript:AT1G13607.1.mrna1:192-260(-)
MQLDQWLVALWVKSHGMHLQVP*
>transcript:AT1G13607.1.mrna1.p11 GENE.transcript:AT1G13607.1.mrna1~~transcript:AT1G13607.1.mrna1.p11 ORF type:complete len:18 (-),score=1.79 transcript:AT1G13607.1.mrna1:142-195(-)
MIAFAIAIRTMPTCNVV*
>transcript:AT1G13607.1.mrna1.p12 GENE.transcript:AT1G13607.1.mrna1~~transcript:AT1G13607.1.mrna1.p12 ORF type:complete len:14 (+),score=1.40 transcript:AT1G13607.1.mrna1:289-330(+)
MYSFVCNAKENKT*
As you can see - all very short peptides, none of which is the result
detected by the simple ORF finder.
Could you please help me understand why are these results missing from my
output and possibly what can be done to obtain them with TransDecoder? Were
they actually missed, or were they filtered for some reason (labeled as low
quality)?
Two thing I noticed:
1. The "correct" ORF starts in position 1 of the transcript (i.e no
UTR) - is this expected to affect TransDecoder's prediction?
2. In TransDecoder's results, I see peptides p8,p10,p11,p12,p18 -
where are all others (e.g. p1-p7)?
Thanks a lot!
—
You are receiving this because you are subscribed to this thread.
Reply to this email directly, view it on GitHub
<#114>, or unsubscribe
<https://github.com/notifications/unsubscribe-auth/ABZRKX24RK4SPK4ERQEHFITSIBOVBANCNFSM4R4KCWXQ>
.
--
--
Brian J. Haas
The Broad Institute
http://broadinstitute.org/~bhaas <http://broad.mit.edu/~bhaas>
|
{kind=link}
|
Thanks for the quick reply. I tried with Running with |
|
It should be reported for the long-orf output. If you're running just a
single transcript through TransDecoder, it's not going to predict
correctly, as it's supposed to run on a full transcriptome - where it
trains its codon usage statistics.
…On Mon, Sep 28, 2020 at 11:13 AM Lior Glick ***@***.***> wrote:
Thanks for the quick reply. I tried with -m 50, which indeed changed the
output, but I'm still not getting the expected peptide. I now get a single
peptide:
>transcript:AT1G13607.1.mrna1.p3 GENE.transcript:AT1G13607.1.mrna1~~transcript:AT1G13607.1.mrna1.p3 ORF type:complete len:50 (+),score=2.34 transcript:AT1G13607.1.mrna1:77-226(+)
MNLIWTLYPNPENTNVKLTWIVQTTLHVGIVLIAIANAIMEPVNACHDS*
Running with -m 70 resulted in no peptides being predicted for this
transcript.
I suspect something else is going on. Any idea what that might be?
—
You are receiving this because you commented.
Reply to this email directly, view it on GitHub
<#114 (comment)>,
or unsubscribe
<https://github.com/notifications/unsubscribe-auth/ABZRKX67LSJ7HTWXJ6WO6MTSICRYBANCNFSM4R4KCWXQ>
.
--
--
Brian J. Haas
The Broad Institute
http://broadinstitute.org/~bhaas <http://broad.mit.edu/~bhaas>
|
|
I ran everything on the full transcriptome (28,280 transcripts). |
|
oh, ok, if it was run on the whole transcriptome with -m >= 50 and it
didn't predict it, it was because the coding scores were insufficient.
If you include blast matches, you can ensure retrieval of orfs that don't
have sufficient coding score metrics.
best,
~b
…On Mon, Sep 28, 2020 at 12:31 PM Lior Glick ***@***.***> wrote:
I ran everything on the full transcriptome (28,280 transcripts).
Not sure if that's what you meant. Is there additional information
somewhere that can help understand why this ORF was not predicted?
—
You are receiving this because you commented.
Reply to this email directly, view it on GitHub
<#114 (comment)>,
or unsubscribe
<https://github.com/notifications/unsubscribe-auth/ABZRKX6EXFRBBL2EXZW2DQLSIC24RANCNFSM4R4KCWXQ>
.
--
--
Brian J. Haas
The Broad Institute
http://broadinstitute.org/~bhaas <http://broad.mit.edu/~bhaas>
|
|
hi,
Here's the documentation for that step:
https://github.com/TransDecoder/TransDecoder/wiki#including-homology-searches-as-orf-retention-criteria
If you have additional orfs that you want to retain, just be sure they're
in that output file. You could fake some entries for them if you need to.
If they're included in that file, they should be retained.
~b
…On Mon, Sep 28, 2020 at 1:05 PM Lior Glick ***@***.***> wrote:
Thanks, I am guessing you're referring to the --retain_blastp_hits
option, right?
Unfortunately I don't really understand how this option should be used.
Specifically, what blastp run should I make to get the required blast6 file?
—
You are receiving this because you commented.
Reply to this email directly, view it on GitHub
<#114 (comment)>,
or unsubscribe
<https://github.com/notifications/unsubscribe-auth/ABZRKX7RL5FQSBVDR4IKULDSIC65TANCNFSM4R4KCWXQ>
.
--
--
Brian J. Haas
The Broad Institute
http://broadinstitute.org/~bhaas <http://broad.mit.edu/~bhaas>
|
|
Thanks. Did that and it worked fine. |
|
I am running TransDecoder 5.5.0 (installed using conda) to predict plant proteins from transcripts.
I used the commands:
I notice some strange behavior. For example, one of the records in
transcripts.fastais:When I run this sequence through NCBI's ORF finder, I get several peptides, including a 72 AA long one which I assume is correct.

However, relevant predictions from TransDecoder's output look like this:
As you can see - all very short peptides, none of which is the result detected by the simple ORF finder.
Could you please help me understand why are these results missing from my output and possibly what can be done to obtain them with TransDecoder? Were they actually missed, or were they filtered for some reason (labeled as low quality)?
Two thing I noticed:
Thanks a lot!
The text was updated successfully, but these errors were encountered: