TieBrush, TieCov and Sashimi: efficient methods for aggregating and summarizing aligned sequences across large datasets
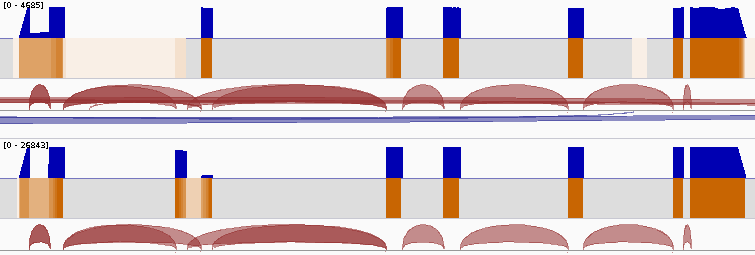
TieBrush is a simple yet efficient method for merging redundant information from multiple alignment files, designed to enable rapid manipulation of extremely large sequencing datasets. The method is specifically designed to optimize investigations of RNA, whole-genome, exome and other types of sequencing experiments. TieBrush preserves much of the original information in a greatly condensed representation as a BAM file, which allows manipulation and extraction of dataset and subset-specific statistics using tools within the package, and which is also compatible with other common utilities.
This utility aims to merge/collapse "duplicate" read alignments (same location with the same CIGAR string), across multiple sequencing samples (multiple input BAM files), adding custom SAM tags in order to keep track of the "alignment multiplicity" count (how many times the same alignment is seen across all input data) and "sample count" (how many samples show that same alignment). The initial goal is to generate this composite BAM file which multiplexes read alignments from many sequencing samples, painting a comprehensive "background" picture of read alignments with their counts across many samples.
Ales Varabyou, Geo Pertea, Christopher Pockrandt, Mihaela Pertea, TieBrush: an efficient method for aggregating and summarizing mapped reads across large datasets, Bioinformatics, 2021;, btab342, https://doi.org/10.1093/bioinformatics/btab342
Sashimi plot is largely based on the implementation from the MISO package. please cite both the TieBrush publication as well as the original MISO paper:
Katz, Yarden, Eric T. Wang, Jacob Silterra, Schraga Schwartz, Bang Wong, Helga Thorvaldsdóttir, James T. Robinson, Jill P. Mesirov, Edoardo M. Airoldi, and Christopher B. Burge. "Quantitative visualization of alternative exon expression from RNA-seq data." Bioinformatics 31, no. 14 (2015): 2400-2402.
If you want to build it from source, we recommend cloning the git repository as shown below to ensure fixed releases of any dependencies are fetched and compiled with the software.
$ git clone https://github.com/alevar/tiebrush.git --recursive
$ cd tiebrush/
$ cmake -DCMAKE_BUILD_TYPE=Release .
$ make -j4
$ make installFor a fully static build -DTIEBRUSH_STATIC_BUILD=1 needs to be added to the list of arguments in the cmake command.
By default make install will likely require administrative privileges. To specify custom installation path -DCMAKE_INSTALL_PREFIX=<custom/installation/path> needs to be added to the list of arguments in the cmake command.
If you are using a very old version of Git (< 1.6.5) the flag --recursive does not exist. In this case you need to clone the submodule separately (git submodule update --init --recursive).
Requirements
- Operating System
GNU/Linux, Mac
- Compiler
GCC ≥ 4.8, LLVM/Clang ≥ 3.8
- Build system
CMake ≥ 2.8
- Language support
C++11
Summarize and filter read alignments from multiple sequencing samples (taken as sorted BAM files). This utility aims to merge/collapse "duplicate" read alignments (same location with the same CIGAR string), across multiple sequencing samples (multiple input BAM files), adding custom SAM tags in order to keep track of the "alignment multiplicity" count (how many times the same alignment is seen across all input data) and "sample count" (how many samples show that same alignment).
The goal is to generate this composite BAM file which multiplexes read alignments from many sequencing samples, painting a comprehensive "background" picture of read alignments with their counts across many samples. :
tiebrush [-h] -o OUTPUT [-L|-P|-E] [-S] [-M] [-N max_NH_value] [-Q min_mapping_quality] [-F FLAGS] ...
Input arguments:
... Input can be provided as a space-delimited list of filenames or as a text file containing a
list of filenames, one per line
Required arguments:
-o File for BAM output
Optional arguments:
-h, --help Show this help message and exit
--version Show the program version end exit
-L, --full If enabled, only reads with the same CIGAR and MD strings will be grouped and collapsed.
By default, TieBrush will consider the CIGAR string only when grouping reads
-P, --clip If enabled, reads will be grouped by clipped CIGAR string. In this mode 5S10M5S and
3S10M3S CIGAR strings will be grouped if the coordinates of the matching substring (10M)
are the same between reads
-E, --exon If enabled, reads will be grouped if their exon boundaries are the same. This option discards
any structural variants contained in mapped substrings of the read and only considers start
and end coordinates of each non-splicing segment of the CIGAR string
-S, --keep-supp If enabled, supplementary alignments will be included in the collapsed groups of reads.
By default, TieBrush removes any mappings not listed as primary (0x100). Note, that if enabled,
each supplementary mapping will count as a separate read
-M, --keep-unmap If enabled, unmapped reads will be retained (uncollapsed) in the output.
By default, TieBrush removes any unmapped reads
-N Maximum NH score (if available) to include.
-Q Minimum mapping quality to include.
-F Bits in SAM flag to use in read comparison. Only reads that have specified flags will be
merged together (default: 0)Note that options -L, -P and -E are mutually exclusive.
- YC:i:N stores the number of alignments that were merged into this alignment record (multiplicity count)
- YX:i:N stores the number of samples that have this alignment (sample count)
- YD:i:N keeps track of the maximum number of contiguous bases preceding the start of the read alignment in the samples(s) that it belongs to. In other words, if the current alignment is part of an exon-overlapping bundle (strand specific!), this value holds the maximum distance from the beginning of the bundle to the start of this alignment, across all samples having this alignment. If the alignment is not in a bundle (i.e. it is preceded by a uncovered region as it is not overlapped by any another alignment with a lower start position), in all the individual samples where that alignment is present, then the
YDvalue is 0 and the tag is omitted from the output file produced by TieBrush. That means that all the alignments lacking aYDtag in the TieBrush output start at the very beginning of an exon-overlapping bundle (i.e. are not overlapped by a preceding alignment with a lower start coordinate).
If either YC or YX tags are missing (i.e. GBamRecord::tag_int() call returns 0) then the alignment is unique (when YC is 0) or only one sample has it (if YX is 0). The actual count in such cases is 1.
The TieCov utility can take the output file produced by TieBrush and can generate the following auxiliary files:
- a BedGraph file with the coverage data (see http://genome.ucsc.edu/goldenPath/help/bedgraph.html); this file can be converted to BigWig (using bedGraphToBigWig) or to TDF format (using igvtools) in order to be loaded in IGV as an additional coverage track
- a junction BED file which can be loaded directly in IGV as an additional junction track (http://software.broadinstitute.org/software/igv/splice_junctions)
- a heatmap BED that uses color intensity to represent the number of samples that contain each position.
tiecov [-s out.sample.bed] [-c out.coverage.bedgraph] [-j out.junctions.bed] [-W] input
Input arguments (required):
input alignment file in SAM/BAM/CRAM format
Optional arguments (at least one of -s/-c/-j must be specified):
-s output BED file with an estimate of the number of samples which contain alignments
for each interval.
-j output BED file with coverage of all splice-junctions in the input file.
-c output BedGraph (or BigWig with '-W') file with coverage for all mapped bases.
-W save coverage to -c file in BigWig format. Default output is in BED format.TieWrap is a small utility script provided to make running TieBrush on large datasets a bit easier. Unlike TieBrush, TieWrap can be launched with as many input files as needed and will automatically divide them into batches processing and combining batches to produce a single representation at the end. All standard TieBrush arguments can be passed over to TieWrap. Additionally size of individual batches as well as the concurrency parameters can be set explicitely.
tiewrap.py [-h] -o OUTPUT [-L|-P|-E] [-S] [-M] [-N MAX_NH] [-Q MIN_MAP_QUAL] [-F FLAGS] [-t THREADS] [-b BATCH_SIZE] ...
Input arguments:
... Input can be provided as a space-delimited list of filenames or as a textfile containing a list of
filenames one per each line.
Required arguments:
-o, --output File for BAM output.
Optional arguments:
-h, --help show this help message and exit
-L, --full If enabled, only reads with the same CIGAR and MD strings will be grouped and collapsed.
By default, TieBrush will consider the CIGAR string only when grouping reads.
-P, --clip If enabled, reads will be grouped by clipped CIGAR string. In this mode 5S10M5S and
3S10M3S cigar strings will be grouped if the coordinates of the matching substring (10M)
are the same between reads.
-E, --exon If enabled, reads will be grouped if their exon boundaries are the same. This option discards
any structural variants contained in mapped substrings of the read and only considers start and
end coordinates of each non-splicing segment of the CIGAR string.
-S, --keep-supp If enabled, supplementary alignments will be included in the collapsed groups of reads. By default,
TieBrush removes any mappings not listed as primary (0x100). Note, that if enabled, each
supplementary mapping will count as a separate read.
-M, --keep-unmap If enabled, unmapped reads will be retained (uncollapsed) in the output.
By default, TieBrush removes any unmapped reads.
-N, --max-nh Maximum NH score of the reads to retain.
-Q, --min-map-qual Minimum mapping quality of the reads to retain.
-F, --flags Bits in SAM flag to use in read comparison. Only reads that have specified flags will be merged
together (default: 0)
-t, --threads Number of threads to use.
-b, --batch-size Number of input files to process in a batch on each thread.
Sashimi.py is a small utility script provided to create vectorized visualizzation of a locus, taking full advantage of the files created by TieBrush suite.
Sashimi plot is largely based on the implementation from the MISO package. please cite both the TieBrush publication as well as the original MISO paper:
Katz, Yarden, Eric T. Wang, Jacob Silterra, Schraga Schwartz, Bang Wong, Helga Thorvaldsdóttir, James T. Robinson, Jill P. Mesirov, Edoardo M. Airoldi, and Christopher B. Burge. "Quantitative visualization of alternative exon expression from RNA-seq data." Bioinformatics 31, no. 14 (2015): 2400-2402.
You must have matplotlib, adjustText and numpy installed to run sashimi.py with python3 which can be installed via
pip3 install matplotlib adjustText numpy
sashimi.py [-h] --gtf GTF [--cov COV] [--sj SJ] -o OUTPUT [--intron_scale INTRON_SCALE]
[--exon_scale EXON_SCALE] [--resolution RESOLUTION] [--fig_width FIG_WIDTH]
[--fig_height FIG_HEIGHT] [--font_size FONT_SIZE] [--nxticks NXTICKS]
[--number_junctions] [--reverse] [--title TITLE [TITLE ...]] [--pickle]
[--compare COMPARE] [--all-junctions]
options:
-h, --help show this help message and exit
--gtf GTF annotation in a GFF/GTF format
--cov COV coverage in bedgraph format or a file containing a list of filenames with coverage
in bedgraph for multiple samples. If a list is provided - the files should be in
the same order as the splice junctions below (if provided)
--sj SJ splice junctions in bed format or a file containing a list of filenames with splice
junctions in bed format for multiple samples. If a list is provided - the files
should be in the same order as the coverage tracks.
-o OUTPUT, --output OUTPUT
Filename for the output figure. The format (png,svg, ...) will be automatically
deduced based on the extension.
--intron_scale INTRON_SCALE
Parameter regulating the scaling of the introns (Default: 20). Decreasing the integer
value will scale introns down in size compared to exons.
--exon_scale EXON_SCALE
Parameter regulating the scaling of the exons (Default: 1). Increasing the integer
value will scale exons down in size compared to introns.
--resolution RESOLUTION
Parameter regulates the smoothing factor of the coverage track (Default: 6). Increasing
the value will increase the smoothing by reducing the number of points on the coverage track.
--fig_width FIG_WIDTH
Width of the figure in inches (Default: 20).
--fig_height FIG_HEIGHT
Height of the figure in inches (Default: 10).
--font_size FONT_SIZE
Size of the font (Default: 18)
--nxticks NXTICKS Number of positional markers to include on the x-axis with labels (Default: 4).
--number_junctions Disables labels idicating coverage of splice junctions
--reverse Flips image horizontally, which is equivalent to setting strand to the opposite value.
--title TITLE [TITLE ...] Title of the figure.
--pickle Save a pickle alongside the figure which can be loaded into a separate instance of
matplotlib for modification.
--compare COMPARE Users can specify one of the input transcripts to serve as a reference. If set, all
transcripts in the input will be compared to the reference and plotted using a dedicated
color pallete. The comparison will visualize in-frame and out-of-frame positions as well
as any intervals missing and extra between the reference and each query transcript
--all-junctions Will force the script to display all junctions, including those not present in the GTF