Rapid and accurate projection of scRNA-seq data onto a curated atlas of Bone Marrow hematopoiesis with balanced representation of HSPCs and differentiated cells.
We provide a large-scale reference atlas of human hematopoiesis with balanced representation of CD34+ stem and progenitor cells together with terminally differentiated populations. Our reference map of 263,159 hematopoietic cells provides us with deep portraits of lineage transitions in both early and late stages of human hematopoiesis.
HSPC annotations within this atlas were carefully validated to maximize concordance with immunophenotypically pure populations established through decades of functional studies. In particular, our transcriptional HSC state is highly concordant with functional LT-HSCs.
We provide functions to rapidly and accurately map query scRNA-seq profiles of either normal or leukemic hematopoietic cells onto our bone marrow atlas. Mapping, QC Filtering, CellType prediction, Pseudotime prediction, and Composition analysis can be performed from raw count matrices within minutes (~10min total for ~10,000 cells on a personal laptop).
To cite this package or learn more, please refer to the bioRxiv pre-print: https://www.biorxiv.org/content/10.1101/2023.12.26.573390
# install dependencies
if(!require(BiocManager, quietly = TRUE)) install.packages("BiocManager")
BiocManager::install(c("AUCell", "doMC"))
if(!require(devtools, quietly = TRUE)) install.packages("devtools")
devtools::install_github("jaredhuling/jcolors") # dependency that is no longer on CRAN
# install BoneMarrowMap package
devtools::install_github('andygxzeng/BoneMarrowMap')
We provide an in-depth tutorial notebook for downloading the reference object and an example query dataset (Roy et al, Cell Rep 2021) and performing reference mapping, celltype classification, pseudotime prediction, and composition analysis on the example query dataset.
You can substitute the example dataset for your own scRNA-seq data. All you need is a raw or ambient RNA-corrected count matrix and cell annotations, ideally within a seurat object. Your data should be pre-filtered by n_counts, n_features, and percent_mito before mapping.
Query mapping is effective across sequencing technologies and hematopoietic tissues. Limitations include lack of coverage of mature Neutrophils (due to limitations in 10x technology) and T-cell precursors (due to tissue source).
To run the tutorial within R markdown, you can download the Rmd file directly from your R console and get started on the tutorial:
# Download R Notebook with mapping tutorial
download.file('https://raw.githubusercontent.com/andygxzeng/BoneMarrowMap_Extras/main/BoneMarrowMap_Tutorial.Rmd',
destfile = 'BoneMarrowMap_Tutorial.Rmd')
We have also used this tool extensively for the mapping of leukemic blasts spanning all various acute and chronic leukemias, except T-ALL. Classification of leukemia depends on lineage-specific features identified by morphology and immunophenotyping. Projection of individual leukemic blasts based on thousands of transcriptomic lineage markers enables precise mapping of cellular states beyond what can be achieved in the clinic today.
After completing the standard tutorial, please see the leukemia projection tutorial to download an example dataset of three diverse AMLs and perform mapping, classification, pseudotime prediction, and composition analysis. This tutorial covers special considerations in mapping QC for leukemia samples and provides additional functions to score leukemia cells for functionally validated LSC signatures.
To run the leukemia mapping tutorial within R markdown, you can download the Rmd file directly from your R console:
# Download R Notebook with mapping tutorial for leukemic samples
download.file('https://raw.githubusercontent.com/andygxzeng/BoneMarrowMap_Extras/main/LeukemiaProjection_Tutorial.Rmd',
destfile = 'LeukemiaProjection_Tutorial.Rmd')
Through a few chunks of code, we will:
- Download and visualize the annotated Bone Marrow Reference Atlas
- Download and load a query dataset of CD34+ HSPCs from Roy et al 2021
- Map the query dataset and filter out low-quality cells
- Predict Hematopoietic Cell Type and Pseudotime score for each query cell
- Visualize projection results and save the mapping results
library(Seurat)
library(tidyverse)
library(symphony)
library(ggpubr)
library(patchwork)
library(BoneMarrowMap)
# Set directory to store projection reference files
projection_path = './'
# Download Bone Marrow Reference - 344 Mb
curl::curl_download('https://bonemarrowmap.s3.us-east-2.amazonaws.com/BoneMarrow_RefMap_SymphonyRef.rds',
destfile = paste0(projection_path, 'BoneMarrow_RefMap_SymphonyRef.rds'))
# Download uwot model file - 221 Mb
curl::curl_download('https://bonemarrowmap.s3.us-east-2.amazonaws.com/BoneMarrow_RefMap_uwot_model.uwot',
destfile = paste0(projection_path, 'BoneMarrow_RefMap_uwot_model.uwot'))
# Load Symphony reference
ref <- readRDS(paste0(projection_path, 'BoneMarrow_RefMap_SymphonyRef.rds'))
# Set uwot path for UMAP projection
ref$save_uwot_path <- paste0(projection_path, 'BoneMarrow_RefMap_uwot_model.uwot')
# Visualize Bone Marrow reference
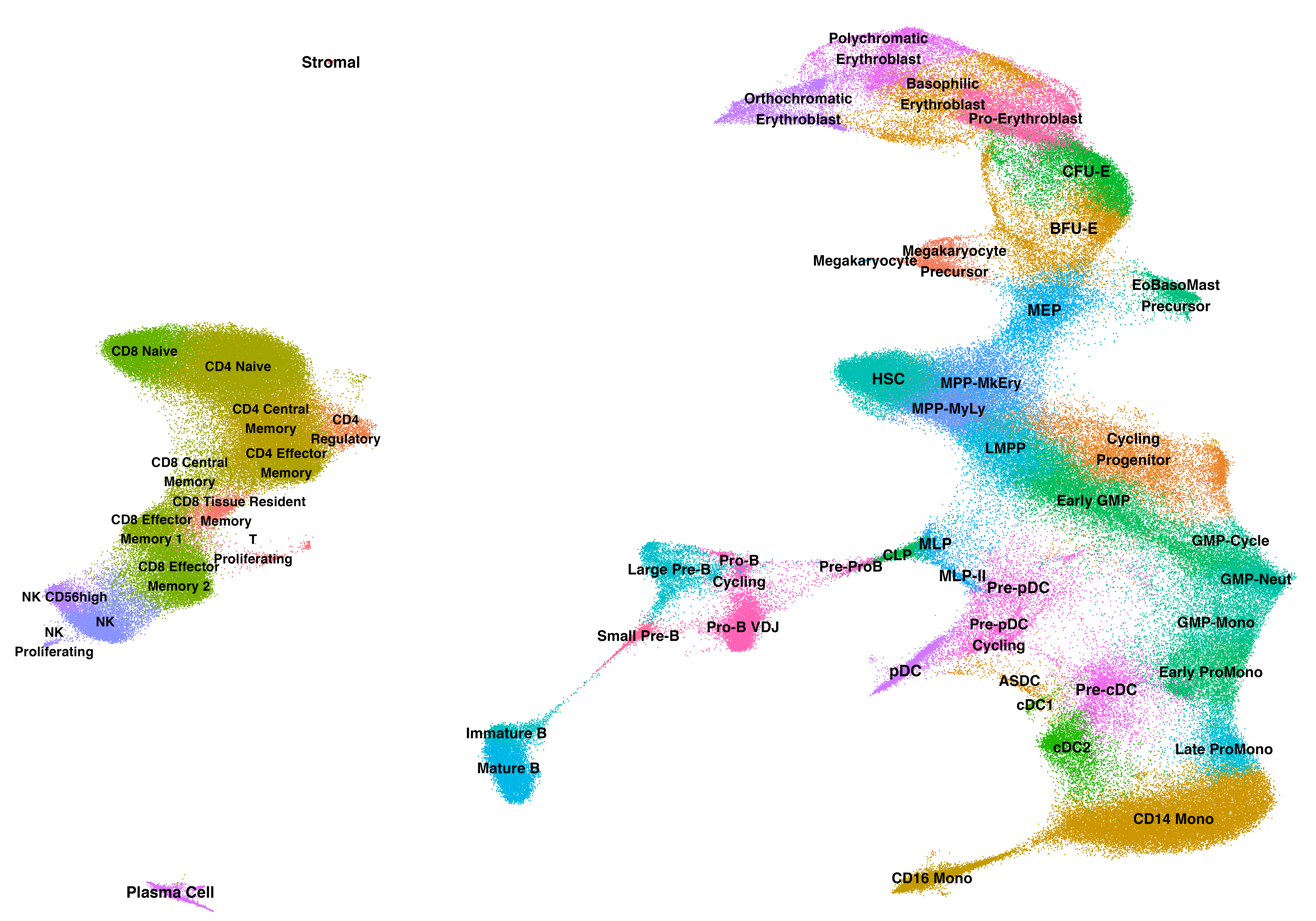
ReferenceSeuratObj <- create_ReferenceObject(ref)
DimPlot(ReferenceSeuratObj, reduction = 'umap', group.by = 'CellType_Annotation_formatted', raster=FALSE, label=TRUE, label.size = 4)
You can use your own query dataset, suggested format is a Seurat object with raw counts
# Load example data from Roy et al - 141 Mb
curl::curl_download('https://bonemarrowmap.s3.us-east-2.amazonaws.com/ExampleQuery_Roy2021.rds',
destfile = paste0(projection_path, 'ExampleQuery_Roy2021.rds'))
query <- readRDS(paste0(projection_path, 'ExampleQuery_Roy2021.rds'))
query
# batch variable to correct in the query data, set as NULL if no batches in query
batchvar <- 'sampleID'
# Map query dataset using Symphony
query <- map_Query(
exp_query = query@assays$RNA@counts,
metadata_query = query@meta.data,
ref_obj = ref,
vars = batchvar
)
# Run QC based on mapping error score, flag cells with mapping error >= 2.5 MADs above median
query <- query %>% calculate_MappingError(., reference = ref, MAD_threshold = 2.5)
plot_MappingErrorQC(query)
# Predict Hematopoietic Cell Types by KNN classification
query <- predict_CellTypes(
query_obj = query,
ref_obj = ref,
final_label = 'predicted_CellType' # celltype assignments with map QC failing cells assigned as NA
)
DimPlot(subset(query, mapping_error_QC == 'Pass'), reduction = 'umap', group.by = c('predicted_CellType'), raster=FALSE, label=TRUE, label.size = 4)
# Predict Pseudotime values by KNN
query <- predict_Pseudotime(
query_obj = query,
ref_obj = ref,
final_label = 'predicted_Pseudotime'
)
FeaturePlot(subset(query, mapping_error_QC == 'Pass'), features = c('predicted_Pseudotime'))
# Set batch/condition to be visualized individually
batch_key <- 'sampleID'
# returns a list of plots for each donor from a pre-specified batch variable
projection_plots <- plot_Projection_byDonor(
query_obj = query,
batch_key = batch_key,
ref_obj = ref,
save_folder = 'projectionFigures/'
)
# show plots together with patchwork
patchwork::wrap_plots(projection_plots)
save_ProjectionResults(
query_obj = query,
file_name = 'querydata_projected_labeled.csv')