{kind=link}
→ bHLH_annotator is also available on the BioInfToolServer
The bHLH_annotator allows the automatic identification and functional annotation of the bHLH transcription factor family in novel plant sequence data sets. Coding sequences or peptide sequences derived from a de novo genome and transcriptome assembly can be analyzed with this pipeline.
A phylogenetic approach is performed for the annotation of the candidates, based on a bait collection of bHLHs and outgroup sequences (non-bHLHs with a high sequence similarity to bHLHs):
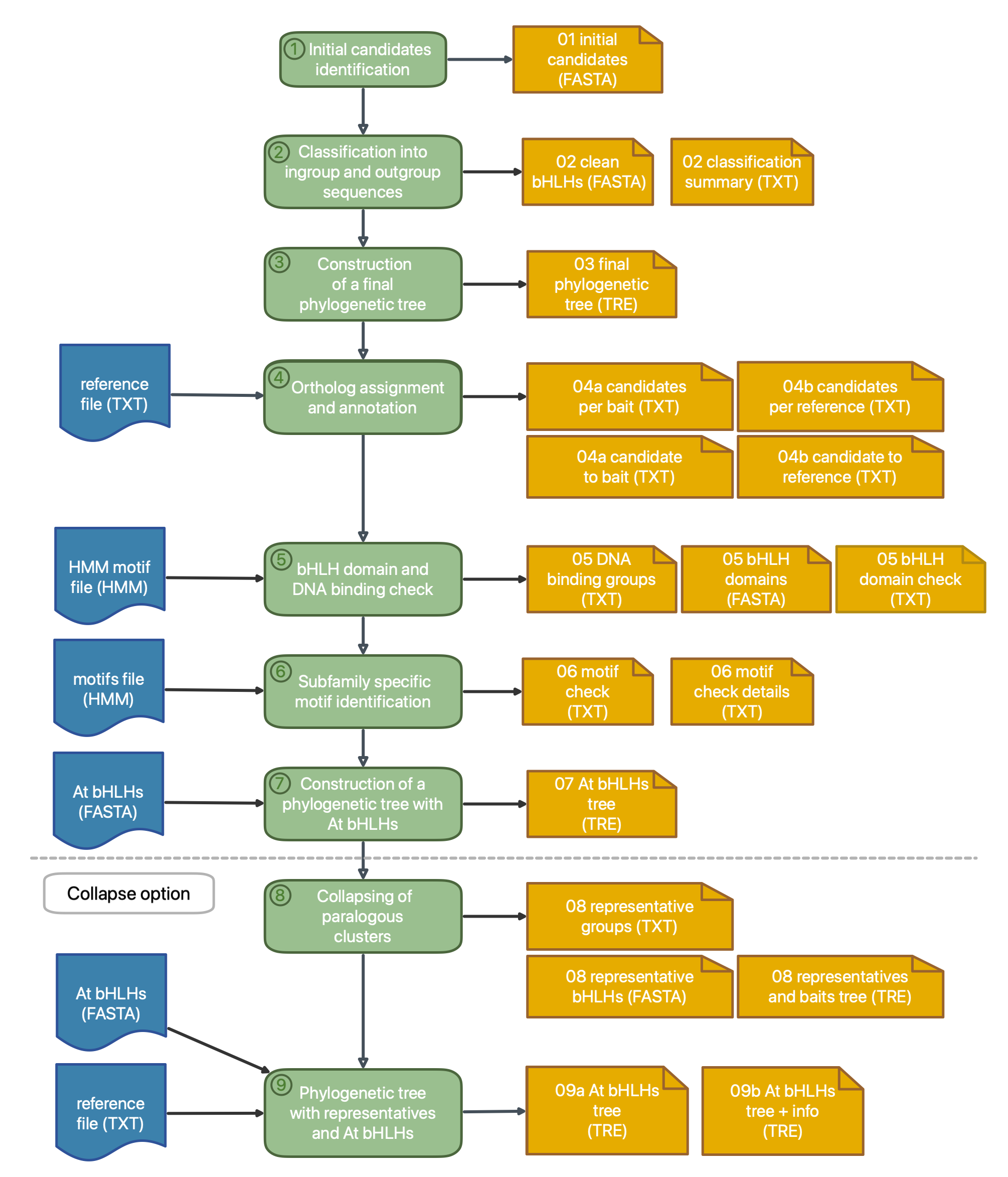
For the identification of initial bHLH candidates (step 1), two search options are available:
- BLAST option (default): Candidates are identified based on sequence similarity to the bait collection. This option is recommended if also bHLHs with a lost domain should be identified.
- HMMER option: Candidates which harbour the HMM motif of the bait collection are identified. This includes candidates with a high specification, that are not represented by the bait collection.
The initial candidates are sorted out based on their phylogenetic relationship to the bHLH and outgroup baits (step 2). The functional annotation of the candidates is assigned by identifying ortholog reference sequences (step 4). As default references, annotated A. thaliana bHLHs are used. Further, bHLH-specific characteristics are analyzed: Presence of the bHLH domain (step 5), DNA-binding properties (step 5), and the identification of subfamily specific motifs (step 6). A phylogenetic tree is constructed with A. thaliana bHLHs to allow a detailed investigation on the foundation of a well-studied species (step 7).
For large datasets like de novo transcriptome assemblies, the collapse option is recommended (step 8 and 9) which collapses paralogous groups by defining a representative candidate. The parallel option is also recommenced to reduce the pipeline runtime and consumption of memory resources during classification.
The data files used in each step can be customised by the user to allow an investigation suiting the own research purpose (described below). A more detailed description of the pipeline and the bait collection can be found here.
The easiest way for installation is by creating an conda environment with the dedicated environment.yml file. This automatically installs all dependencies.
git clone https://github.com/bpucker/bHLH_annotator
cd bHLH_annotator
conda env create -f environment.yml
conda activate bHLH_annotatorThe following dependencies are necessary for the execution of the pipeline:
- Python3:
sudo apt install python3.11(other versions are also compatible)- dendropy:
sudo apt install python3-pip && python3 -m pip install -U dendropy- pandas:pip install pandas- numpy:pip install numpy- matplotlib:pip install matplotlib
- dendropy:
- BLAST:
sudo apt install ncbi-blast+ - HMMER:
conda install -c bioconda hmmer - MAFFT :
sudo apt install mafft - Muscle5 (precompiled binaries recommended)
- FastTree2:
sudo apt-get install -y fasttree - RAxML-NG (precompiled binaries recommended)
The bHLH_annotator can be cloned from github:
git clone https://github.com/bpucker/bHLH_annotator
cd bHLH_annotatorThe pipeline is executed through the following command:
cd <PATH>/bHLH_annotator
python3 bHLH_annotator.py --subject <PATH> --out <OUTPUT> --info <DEFINITION_FILE>
The --subject file defines the path to the input FASTA file containing coding or peptide sequences. The output directory is defined with the --out command. In the output directory, a RESULT folder is created containing the output files created in the pipeline steps. The --info file represents the bHLH_annotator.csv. This file is necessary as it defines the input data files utilized in the pipeline.
| Command | Description | Default |
|---|---|---|
--name <STR> |
Prefix of output file names | -- |
--cdsinput |
Changes expected input to CDS | -- |
--keepnames |
Prevents splitting of sequence names at first space | -- |
--collapse |
Reduces paralogs to one representative | -- |
--parallel |
Parallel option for classification | -- |
| Command | Description | Default |
|---|---|---|
--search <STR> |
Search option for the initial search (blast / hmmer) |
blast |
--mode_aln <STR> |
Alignment tool (muscle / mafft) |
muscle |
--mode_tree <STR> |
Tool for tree construction (fasttree / raxml) |
fasttree |
--blastp <STR> |
Path to blastp | blastp |
--makeblastdb <STR> |
Path to makeblastdb | makeblastdb |
--hmmsearch <STR> |
Path to hmmsearch | hmmsearch |
--mafft <STR> |
Path to MAFFT | mafft |
--muscle <STR> |
Path to muscle | muscle |
--fasttree <STR> |
Path to FastTree | fasttree |
--raxml <STR> |
Path to RAxML | raxml-ng |
| Command | Description | Default |
|---|---|---|
--bitcutp <INT> |
BLASTp bitscore cutoff | 60 |
--simcutp <INT> |
BLASTp similarity cutoff | 40.0 |
--poscutp <INT> |
Max number of BLASTp hits per bait | 100 |
--lencutp <INT> |
Min BLASTp alignment length | 80 |
--filterdomain |
Filter candidates not matching the HMM motif of the bait collection | -- |
--minscore <FLOAT> |
Minimal score to be considered as ingroup | 0.5 |
--numneighbours <INT> |
Neighbours to consider for classification | 10 |
--neighbourdist <FLOAT> |
X*average nearest neighbour distance is used as minimal distance cutoff to be considered as a neighbour | 5 |
--minneighbours <INT> |
Minimal number of bait neighbours to be considered as ingroup | 0 |
--paralogdist <FLOAT> |
X*average nearest neighbour distance is used as cutoff to identify paralogs | 10.0 |
--numprocesscandidates <INT> |
Number of candidates processed at the same time in the parallel option | 200 |
| Command | Description | Default |
|---|---|---|
--cpu <INT> |
Number of threads | 4 |
--cpumax <INT> |
Maximal number of threads for classification (step 2) | value of --cpu |
--cpub <INT> |
Number of threads for BLASTp search (step 1) | value of --cpu |
--cpur <INT> |
Number of threads for alignment/tree construction | value of --cpu |
The data input files required as resources for the steps of the pipeline are defined in the bHLH_annotator.csv file. The default files are stored in the data folder. The following files are required as resources:
| Argument | File | Description |
|---|---|---|
--baits <PATH> |
Baits | Contains the bHLH and outgroup sequences of the bait collection (mandatory) |
--baitsinfo <PATH> |
BaitsInfo | Info file defining each bait as bHLH or outgroup sequence (mandatory) |
--optimisedbaits <PATH> |
OptimisedBaits | Optimised bait collection containing only phylogenetic distinct baits that are used for tree construction (recommended) |
--reference <PATH> |
Reference | References with alternative name, functional annotation and subfamily (step 4 and 9) |
--hmm <PATH> |
HMM | HMM motif representing the bHLH domain (step 2 for HMMER search, step 5, and --filterdomain option) |
--motifs <PATH> |
Motifs | HMM motifs of subfamily specific motifs (step 5) |
--ath <PATH> |
Ath | A. thaliana sequences used for tree construction (step 7 and 9) |
The defined files must meet the following requirements:
- Baits and OptimisedBaits: All baits, including optimised baits, must be categorized in the BaitsInfo file as ingroup or outgroup sequences.
- BaitsInfo: All sequences categorized in the BaitsInfo file must be included in the Baits file.
- Reference: The sequences of the specified references must be included in the Ath sequence file or the bait collection.
If the optional files are not defined in the bHLH_annotator.csv file or via argument, the dependent pipeline steps are skipped. If the files are defined both in the csv file and via argument, the argument is prioritized. The files refered to need to be placed in the 'data' folder or defined using complete paths.
Python, dendropy, pandas, numpy, matplotlib, BLAST+, HMMER, MAFFT or MUSCLE5, FastTree2 or RAxML
Thoben C. and Pucker B. (2023). Automatic annotation of the bHLH gene family in plants. BMC Genomics 24, 780 (2023). doi: 10.1186/s12864-023-09877-2.