Using SamTools to view alignments
SamTools is a set of tools for working with alignments. Bowtie is used in methylcode and can output alignments in SAM format, since we must convert and flip the reads and reference genome for the bisulfite alignment, the resulting output is not immediately useful. However, when methylcode is used to align BS treated reads to a reference genome, it also creates an additional SAM alignment file where the positions and strand information is corrected. This file is created in the output directory and named: methylcoded.sam.
In order to view the BS treated reads that align to a reference sequence, one can follow a few simple steps.
note as of version 0.2.3 methylcoder will output a shell script to outdir/commands.sam.sh that will perform the preparatory steps described below.
the following is the transcript of a shell session of the commands which end with viewing the reads aligned to the genome in the sam-tools tview tool. Here out is the directory where the methylcode output is sent. Note you must create a chr.lengths.txt file which has the format [chr]\t[length] to specify the length for each chromosome.
$ cd out
$ ls -lh methylcoded.sam
-rw-r--r-- 1 brentp brentp 6.9G 2010-03-09 17:42 methylcoded.sam
Convert a .sam file to .bam the file—specified here as chr.lengths.txt . Then sort. Then create an index.
$ ~/src/samtools/samtools view -b -t chr.lengths.txt methylcoded.sam -o methylcoded.unsorted.bam
$ ~/src/samtools/samtools sort methylcoded.unsorted.bam methylcoded # .bam suffix is added automatically
$ ~/src/samtools/samtools index methylcoded.bam
create a fasta where we’re sure that it doesn’t have long lines (sam-tools doesnt like those).
note you only ever need to do this once per reference genome, or not at all if your fasta is already
wrapped/folded. Once it’s wrapped, sam-tools can index it.
$ fold /labdata/thaliana_v9/thaliana_v9.fasta > /labdata/thaliana_v9/thaliana_v9.fold.fasta
$ ~/src/samtools/samtools faidx /labdata/thaliana_v9/thaliana_v9.fold.fasta
then you can view the alignments in the sam-tools viewer:
$ ~/src/samtools/samtools tview methylcoded.bam /labdata/thaliana_v9/thaliana_v9.fold.fasta
From there, you can *g*o to a particular location by typing `g` then a chromosome:location e.g.
1:100,000
And read more about the viewer here or press ‘?’ in the viewer for help.
you should get something like this
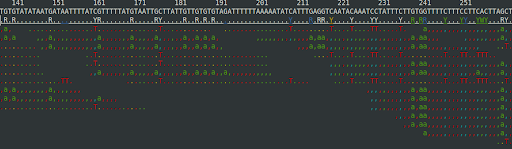
Notice that—for the most part—only the T’s and a’s are mismatching. The a’s are lowercased—indicating that they are from reads matching the reverse strand (G converted to A equates to C to T after complementing).