{kind=link}
{kind=link}
{kind=link}
Convert Dynamo table, relion 4.0 (3.1) star file to script to visualize in ChimeraX Now can do in batch instead of manually doing every tomogram.
Please cite subtomo2Chimera code repository DOI 10.5281/zenodo.6820119
starfile and eulerangles modules from Alister Burt
If you use dynamotable2ChimeraX.py, you need to also install dynamotable
pip install eulerangles
pip install starfile
pip install dynamotable
python relionsubtomo2ChimeraX.py --i run_data_empiar10064.star --o load_tomo1.cxc --avgAngpix 10.48 --avgBoxSize "64,64,64" --tomoname CTEM_tomo1
For Relion 3.1
python relionsubtomo2ChimeraX.py --i run_data_empiar10064.star --o load_tomo1.cxc --avgAngpix 10.48 --avgBoxSize "64,64,64" --tomoname CTEM_tomo1 --relion31 1
--i Input star file
--o Output ChimeraX Loading Script
--avgAngpix Pixel Size of the average file
--avgBoxSize Box Size of the average. 3 values seperated by comma and double quoted. It is ok to use non-cube as long as same center as the cubic subtomogram.
--tomoname Name of tomo for the visualization. You have to do this script for each tomo you want to visualize. And the tomo name needs 100% match.
Not required option
--avgFileName Name of subtomo average loaded for the visualization. Default is avg.mrc but you can put any name in.
--offset (default 0) Offset number if you want to load different kinds of subtomogram averages into the same session
--level (default 0.0039) Volume level set for the subtomogram average
--relion31 (default 0) Relion 3.1 or Relion 4.0 (1 or 0 value).
python dynamotable2ChimeraX.py --tomoDoc tomograms.doc --i aligned.tbl --o load_tomo.cxc --avgAngpix 8.48 --tomoAngpix 8.48 --avgBoxSize "128,128,15" --tomoname CU428_TS013_rec.mrc
--i Input table file
--tomoDoc Input tomo doc file
--o Output ChimeraX Loading Script
--avgAngpix Pixel Size of the average file
--avgBoxSize Box Size of the average. 3 values seperated by comma and double quoted. It is ok to use non-cube as long as same center as the cubic subtomogram.
--tomoname Name of tomo for the visualization. You have to do this script for each tomo you want to visualize. Name needs 100% match
--tomoAngpix Pixel size of the tomogram used for Dynamo table.
Not required option
--avgFileName Name of subtomo average loaded for the visualization. Default is avg.mrc but you can put any name in.
--offset (default 0) Offset number if you want to load different kinds of subtomogram averages into the same session
--level (default 0.0039) Volume level set for the subtomogram average
python batchrelionsubtomo2ChimeraX.py --i run_data_empiar10064.star --o load.cxc --avgAngpix 10.48 --avgBoxSize "64,64,64"
For Relion 3.1
python batchrelionsubtomo2ChimeraX.py --i run_data_empiar10064.star --o load.cxc --avgAngpix 10.48 --avgBoxSize "64,64,64" --relion31 1
python batchdynamotable2ChimeraX.py --tomoDoc tomograms.doc --i aligned.tbl --o load.cxc --avgAngpix 8.48 --tomoAngpix 8.48 --avgBoxSize "128,128,15"
For batch option The same as before without giving the --tomoname
The script will load the subtomogram average in ChimeraX and transform it to orientation of each subtomo in the tomogram. You can also load the tomogram in to visualize it together. This script only perform for only one kind of subtomogram average but it is not too difficult to do the same thing with different classes of average using the offset and combine the loading script together.
The subtomogram average name is avg.mrc by default. You need to adjust the level of the average in output ChimeraX loading script. You can use the exact command above to test the example of ribosome subtomo average from EMPIAR-10064.
The output ChimeraX loading script uses a lot of memory due to many subtomograms rendering. Therefore, try to make the avg.mrc file as small as possible. It doesn't have to be the same pixel size as the star file data. You can use further binning/tight crop and then input the pixel size & box size of the subtomogram average into the command
Update: v0.1 2022/05/03 Now you can customize the subtomo avg name & the offset is now correct. Update: v0.11 2022/06/08 Fix the index for non-sorted star file. Also can use --relion31 flag for doing Relion3.1 file directly.
NOTE 1:
For calculation of rotation matrix: The rotation matrix needs to be transposed due to the view orientation of ChimeraX (turn x 180).
The shift after the rotation = RotationMatrix*-HalfBoxSize + CoordinateOfSubtomogram
NOTE 2: Script only works with relion 4.0 star file.
-
Prepare the input files: Relion star file from Refine3D job (Should be at least 3.1 and up), the binned out subtomogram average avg.mrc (size ~1Mb). For Dynamo, table & tomogram doc file, the binned out subtomogram average avg.mrc.
-
Generate the ChimeraX loading script for a specific tomogram using the above command
python relionsubtomo2ChimeraX.py --i run_data_empiar10064.star --o load_tomo1.cxc --avgAngpix 10.48 --avgBoxSize "64,64,64" --tomoname CTEM_tomo1*
Full command
python relionsubtomo2ChimeraX.py --i run_data_empiar10064.star --o load_tomo1.cxc --avgAngpix 10.48 --avgBoxSize "64,64,64" --tomoname CTEM_tomo1 --level 0.0039 --avgFileName avg.mrc --offset 0*
For dynamo command
python dynamotable2ChimeraX.py --tomoDoc tomograms.doc --i aligned.tbl --o load_tomo.cxc --avgAngpix 8.48 --tomoAngpix 8.48 --avgBoxSize "128,128,15" --tomoname CU428_TS013_rec.mrc
- Edit the ChimeraX loading script for proper level of the map and open the output script in Chimera using commandline or interface:
chimerax load_tomo1.cxc
-
Loading the tomogram into ChimeraX (Better bin4 or more for faster loading)
-
Set the right pixel size and origin for the tomogram to be properly located relative to the subtomogram average
volume #TomoModelNumber voxelSize 10.48 originIndex 0,0,0
- One you have the load_tomo1.cxc from above, you can actually use stl file for faster loading time or flexible coloring as well. First, you need to convert the surface render of avg.mrc to a STL file. Load avg.mrc in ChimeraX, set a proper threshold like you want to see it. Then save the surface rendering as STL file
save avg.stl format stl models #1
- Regenerate the loading cxc script
python relionsubtomo2ChimeraX.py --i run_data_empiar10064.star --o load_tomo1_stl.cxc --avgAngpix 10.48 --avgFileName avg.stl --avgBoxSize "64,64,64" --tomoname CTEM_tomo1
View of the subtomogram average only

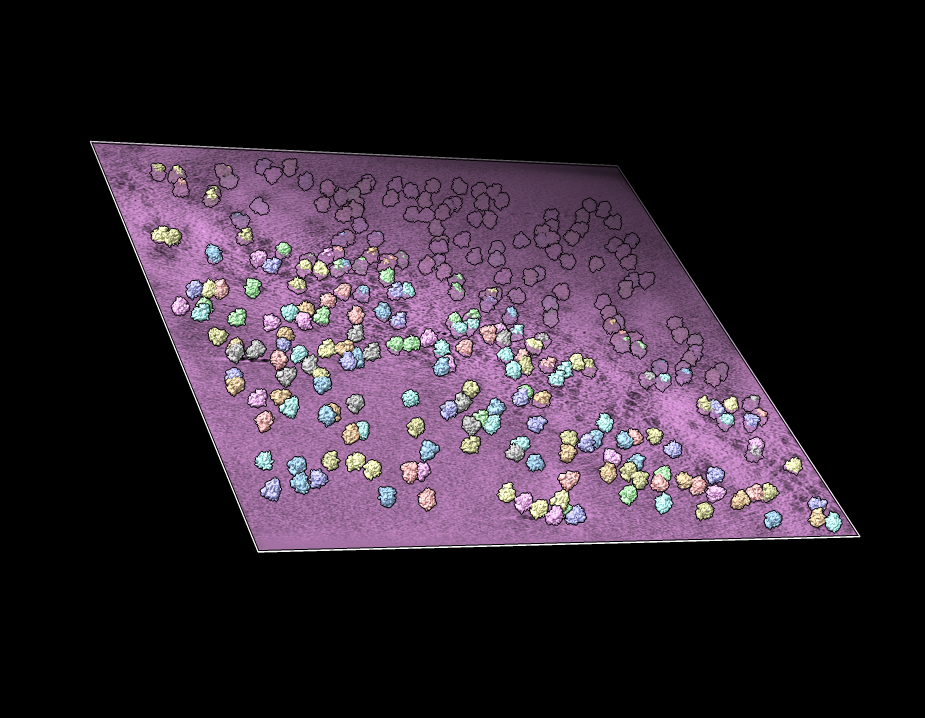
View of subtomogram average within the tomogram

Plane View