Home
Before you start, please README.
Short reads mapped to a reference genome.
Must contain the reference sequence a user wants to analyze.
Must contain the reference sequence a user wants to analyze and have coding regions annotated.
samtools view -H bam_file.bam | grep ^@SQ | grep 'seq_id'
grep ">seq_id" fasta_file.fasta
grep "seq_id" gff_file.gff
Note: The sequence id must be the same in all files and must be exact as input provided by a user.
- K03455.gff
- Must contain CDS annotation
samtools view -H bam_file.bam | grep ^@SQ | grep 'K03455|HIVHXB2CG'- Output:
@SQ SN:K03455|HIVHXB2CG LN:9719
grep "K03455|HIVHXB2CG" gff_file.gff- Output:
K03455|HIVHXB2CG 1 9719 - Multiple lines are possible
- Must check that it contains CDS annotation and has region a user is interested in
grep "K03455|HIVHXB2CG" fasta_file.fasta- Output:
>K03455|HIVHXB2CG xxyy dtdt - Only one line is expected as output
- After space information vary based on annotation in fasta
- Basic command:
seqpanther codoncounter -bam data/hiv/bam/Ko48924_K03455_HIVHXB2CG.bam -rid K03455\|HIVHXB2CG -ref data/hiv/K03455.fasta -gff data/hiv/K03455.gff - The command will generate 3 files in the current directory:
codon_output.csv,indel_output.csvandsub_ouput.csv - For more options, run
seqpanther codoncounter -hor visit SeqPather Repo - Details on the outputs are also available in the README at SeqPather Repo
- Command:
seqpanther cc2ns -s sub_output.csv -i indel_output.csv - The command will generate one file per bam (tsv format). File name will start with bam prefix (before .bam section).
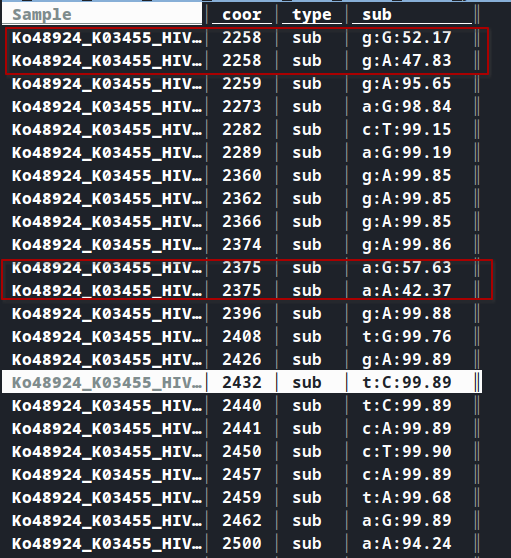
- Coordinate
2258and2375have multiple type of change. Not required change should be removed. In case of multiple changes, last alteration will be considered.
- Command :
samtools index data/hiv/bam/Ko48924_K03455_HIVHXB2CG.bam - Command :
samtools mpileup -uf data/hiv/K03455.fasta data/hiv/bam/Ko48924_K03455_HIVHXB2CG.bam | bcftools call -c | vcfutils.pl vcf2fq > Ko48924_K03455_HIVHXB2CG.fastq - Command :
seqtk seq seq -aQ64 Ko48924_K03455_HIVHXB2CG.fastq > consensus/Ko48924_K03455_HIVHXB2CG.fasta
- Command:
seqpanther nucsubs -r data/hiv/K03455.fasta -c consensus -t changes -o output -i K03455\|HIVHXB2CG