Note: This repository contains scripts that were developed for "SimSCSnTree: a simulator of single-cell DNA sequencing data".
Authors: Xian Fan (xfan2@fsu.edu), Luay Nakhleh (nakhleh@rice.edu)
- Installing SimSCSnTree.
- Usage of SimSCSnTree.
- Examples.
- Simulating both CNAs and SNVs on a tree (step 1).
- Simulating reads at the DOP-PCR read depth fluctuation (and bulk and MALBAC) (step 2).
- Simulating multiple levels of internal nodes (step 2).
- Simulating clones of cells (step 2).
- Simulating bulk sequencing (step 2).
- Simulating large dataset
- Simulating reads with different ploidies
- Simulating reads with different levels of fluctuation
- Miscellaneous
There are two ways to install SimSCSnTree.
The first is through BioConda. To do this, just use
conda install simscsntree
It may take a while for the metadata to be collected and installed. This option is preferred because you don't have to install all the dependencies or worry about the python version. You can skip Software requirements and Environment setup section except step 2 in Environment setup as wgsim is not available on BioConda. To do that, download the package from this github link (git clone https://github.com/compbiofan/SimSCSnTree.git) and go to the SimSCSnTree directory, further down to wgsim-master subfolder. Follow the direction in step 2 in Environment setup section to install the wgsim. After you are done with installing wgsim, download the reference file directed in the Data requirement section. If you use the BioConda option, use
python -m SimSCSnTree
to launch all the commands.
The second is through downloading the files in github. If that is the case, you have to use
python main.par.overlapping.py
to launch all the commands. In this document, we show the command lines using this option.
A reference file such as hg19.fa, which can be downloaded here.
The following applies if you don't use BioConda to install SimSCSnTree.
-
Python 3 or up.
-
Python modules: numpy 1.18 or above, anytree.
Suppose $this_dir is the path of this package.
-
Add pipeline to your directory. In bash,
if [ -d $this_dir ]; then PATH="$this_dir:$PATH" fi -
Make the binary from the revised wgsim.
cd $this_dir/wgsim-mastergcc -g -O2 -Wall -o wgsim wgsim.c -lz -lmchmod u+x wgsim -
Python modules: numpy, anytree.
pip install numpypip install anytree
SimSCSnTree has two steps. Step 1 generate a tree, each node of which contains a genome and each edge of which CNA(s) and/or SNV(s) are imputed. Step 2 samples reads from the genomes on the nodes selected. The following shows how each step works.
1. Generate the tree with CNVs/SNVs on the edges. This step generates two npy files, one for the tree (containing all information of CNVs/SNVs on the edges and the tree structure) and one for intermediate files (containing chromosome name, chromosome length and the index of the nodes). The .npy files will be used in step 2 for sampling reads.
python main.par.overlapping.py
followed by the following parameters grouped by their functions.
-
Parameters controlling file IO: -r, -t and -o. -r is where all the output data will be stored. Default is "test" under the current path. -t shall be defined to locate the reference file where the reads are sequenced from. -o is a log file with default std.out.
-r (--directory) Location of simulated data. The program will remove the whole directory if it already exists. Otherwise it will create one. (default: test)-t (--template-ref) The reference file to sequence the reads.-o (--outfile) The standard output file, will be saved in output folder, just give the file name. (default: std.out) -
Parameters controlling tree structure: -n, -B, -A, -G, -K, -F and -H. -n is the total number of cells on a level of interest. Since -F is the total number of nodes (subclones) on the leaf level, -n should always be greater than -F unless when -M is 1. When -M is not 1, the cells will be distributed according to their percentage on each node for a certain level. The percentage of a node is specified by the Beta-splitting model when the binary tree grows and this is a stochastic process. -F and -H defines the total number of subclones on the leaf level, thus the width of the tree. -F and -H are the mean and standard deviation that the tree width will be sampled from in a Gaussian distribution. Define -H to be a very small number such as 0.0001 when users want the tree width to be a certain number. The binary tree's branch splitting follows Beta-splitting model so that the splitting of the cells between the left and right branches for each split follows a Beta distribution, whose alpha and beta parameters are specified by -A (--Alpha) and -B (--Beta). When -B and -A are closer to each other (e.g., 0.5 and 0.5), the tree is more balanced. To generate a tree that is unbalanced, make -B and -A be far from each other but still within [0, 1]. -G and -K are the mean and standard deviation of the Gaussian distribution that the depth of the tree (the highest level of the tree) will be sampled from. The splitting ends when the number of nodes on the leaf level reaches the tree width. -G and -K defines the tree's depth. -G and -K are the mean and standard deviation the tree's depth will be drawn from in a Gaussian distribution. The tree grows in a way that prioritizes to reach defined tree depth. But if -G is unreasonably big, e.g., -G > 2 * -F, the tree's depth may not reach the desired tree depth.
-n (--cell-num) Number of the cells. Always greater than -F treewidth. Treewidth controls the total number of clones whereas cell-num controls the total number of cells sequenced at a certain tree depth. (default: 8)-B (--Beta) The program uses the Beta-splitting model to generate the phylogenetic tree. Specify a value between [0, 1]. (default: 0.5)-A (--Alpha) The Alpha in Beta-splitting model. Specify a value between [0, 1]. The closer Alpha and Beta, the more balanced the tree. (default: 0.5).-G (--treedepth) The mean of the tree depth distribution. The final tree depth will be sampled from a Gaussian with this mean and a fixed standard deviation. (default: 4 counting from the first cancer cell)-K (--treedepthsigma) The standard deviation of the tree depth distribution. To get exactly the tree depth defined by -F, use a very small standard deviation, e.g., 0.0001. (default: 0.5)-F (--treewidth) The mean of the tree width distribution. The final tree width will be sampled from a Gaussian with this mean and a fixed standard deviation. (default: 8)-H (--treewidthsigma) The standard deviation of the tree width distribution. To get exactly the tree width defined by -F, use a very small standard deviation, e.g., 0.0001. (default: 0.5) -
Parameters controlling CNAs: -c, -d, -m, -e and -a. To impute a CNA on a branch on the tree, the number of CNAs follows a Poisson distribution, the mean of which follows an exponential distribution with p specified by -c (--cn-num). The deletion rate as compared to copy number gain follows a binomial distribution with p specified by -d (--del-rate). The CNA size follows an exponential distribution with p specified by -e (--exp-theta) plus a minimum CNA size specified by -m (--min-cn-size). If it is a copy number gain, the numbers of gain follows a Geometric distribution with p specified by -a (--amp-p).
-c (--cn-num) The average number of copy number variations to be added on a branch. (default: 1)-d (--del-rate) The rate of deletion as compared to amplification. (default: 0.5)-m (--min-cn-size) Minimum copy number size. (default: 2000,000bp)-e (--exp-theta) The parameter for the Exponential distribution for copy number size, beyond the minimum one. (default: 5000,000bp)-a (--amp-p) The parameter for the Geometric distribution for the number of copies amplified. (default: 0.5) -
Parameters controlling whole chromosome duplication on the branch to the root: -X, -W, -C, -E and -J. The whole chromosome duplications are imputed in the trunk branch connecting the normal cell and the first tumor cell if -W (--whole-amp) is 1. For each chromosome, the probability that it is amplified equals -C (--whole-amp-rate) and the number of copies amplified follows a geometric distribution with p specified by -J (--amp-num-geo-par) multiplied by a number specified by -E (--whole-amp-num).
-X (--multi-root) The multiplier of the mean CNV on root. (default: 4)-W (--whole-amp) If there is whole chromosome amplification, 1 as yes. (default: 1)-C (--whole-amp-rate) Whole amplification rate: rate of an allele chosen to be amplified (default: 0.2)-E (--whole-amp-num) Whole amplification copy number addition, which occurs to one allele at a time. (default: 1)-J (--amp-num-geo-par) Whole amplification copy number distribution (geometric distribution parameter: the smaller, the more evenly distributed). (default: 1) -
Parameters controlling SNVS: -R. On a branch, the number of SNVs imputed follows a Poisson distribution, the mean of which equals to snv-rate (specified by -R) multiplied by branch length which is sampled from an exponential distribution with p=1.
-R (--snv-rate) The rate of the snv. snv-rate * branch-length = # snvs. (default: 1)
2. Sample reads from specified genomes. This is the 2nd step. Given the two .npy files in the 1st step, this step is to sample the reads for each genome of interest.
python main.par.overlapping.py -k 1
followed by the following parameters grouped by their functions.
-
Parameter controlling which step to run: -k. The second step is to sample reads once the genomes are generated and stored in .npy files. To skip the first step and jump to the second step, use -k option. The program will automatically read the .npy files and start sequencing the reads from the genomes stored in .npy files.
-k (--skip-first-step) If the alleles for all nodes have been made, the step can be skipped. Make it 1 then. (default: 0) -
Parameters for IO: -f. This option specifies the prefix string of the fasta file names for the genomes written for sampling reads.
-f (--fa-prefix) The prefix of the alleles and read names. (default: ref) -
Parameter specifying which read simulator to use: -S. This package has the wgsim source files but users are supposed to compile wgsim and specify the binary by -S.
-S (--wgsim-dir) The directory of the binary of wgsim. It is in the same folder of this main.py. (need to specify) -
Parameters controlling read depth fluctuation (-x, -y, -w and -u), read coverage (-v) and read length (-l). Read depth fluctuation is decided by -x (--Lorenz-x) and -y (--Lorenz-y) which represent the x and y values on the Lorenz curve that imitates the read coverage fluctuation. Suppose -x is fixed at 0.5, the lower the -y, the more uneven the read depth is. When -y is around 0.38, it resembles bulk sampling. For more details, please refer to "Assessing the performance of methods for copy number aberration detection from single-cell DNA sequencing data" authored by XFM, ME, NN and LN in 2020. SimSCSnTree divides the genome into nonoberlapping windows and samples the number of reads for each window. To determine window size, use -w (--window-size). The higher the -w, the less change of read depth on the genome. Starting from the first window which was given a fixed read number according to -v, the next window's read number is calculated by -x, -y and -u, whereas -u (--acceptance-rate) is the probability to accept a proposal in Metropolis Hasting so that the read coverage fluctuation over the whole genome reflects the expected Lorenz curve and the change of the number of reads between neighboring windows is restricted. For more details, please refer to "Assessing the performance of methods for copy number aberration detection from single-cell DNA sequencing data" authored by XFM, ME, NN and LN in 2020. The higher -u, the faster the program would run although the difference may not be noticeable. Users can change the coverage and read length of the sampled reads by tuning -v (--coverage) and -l (--readlen), respectively. -v is the average number of reads covering each nucleotide. For high coverage data such as the data suitable for SNV detection, use -v >= 5. For low coverage data such as the data suitable for CNA detection, use -v < 0.1. SimSCSnTree samples paired-end reads and a single read length is specified by -l. Use the number in between 35 and 150 for mimicking Illumina reads' length.
-x (--Lorenz-x) The value on the x-axis of the point furthest from the diagonal on the Lorenz curve imitating the real coverage uneveness. (default: 0.5)-y (--Lorenz-y) The value on the y-axis of the Lorenz curve imitating the real coverage unevenness. x > y. The closer (x, y) to the diagonal, the better the coverage evenness. (default: 0.4)-v (--coverage) The average coverage of the sequence. (default: 0.02)-l (--readlen) Read length for each read sequenced. (default: 35bp)-w (--window-size) Within a window, the coverage is according to a Gaussian distribution. Neighboring windows' read coverage is according to a Metropolis Hasting process. (default: 200000bp)-u (--acceptance-rate) The probability to accept a proposal in Metropolis Hasting. (default: 0.5) -
Parameters for sequencing ancestral nodes: -L and -U. These two are similar options: -L controls the levels to be sequenced for single-cell sequencing and -U controls the levels to be sequenced for bulk sequencing. -U can be "NA", in which case bulk sequencing will not be performed. For -L, if it is not specified, the program by default sequences the leaf cells.
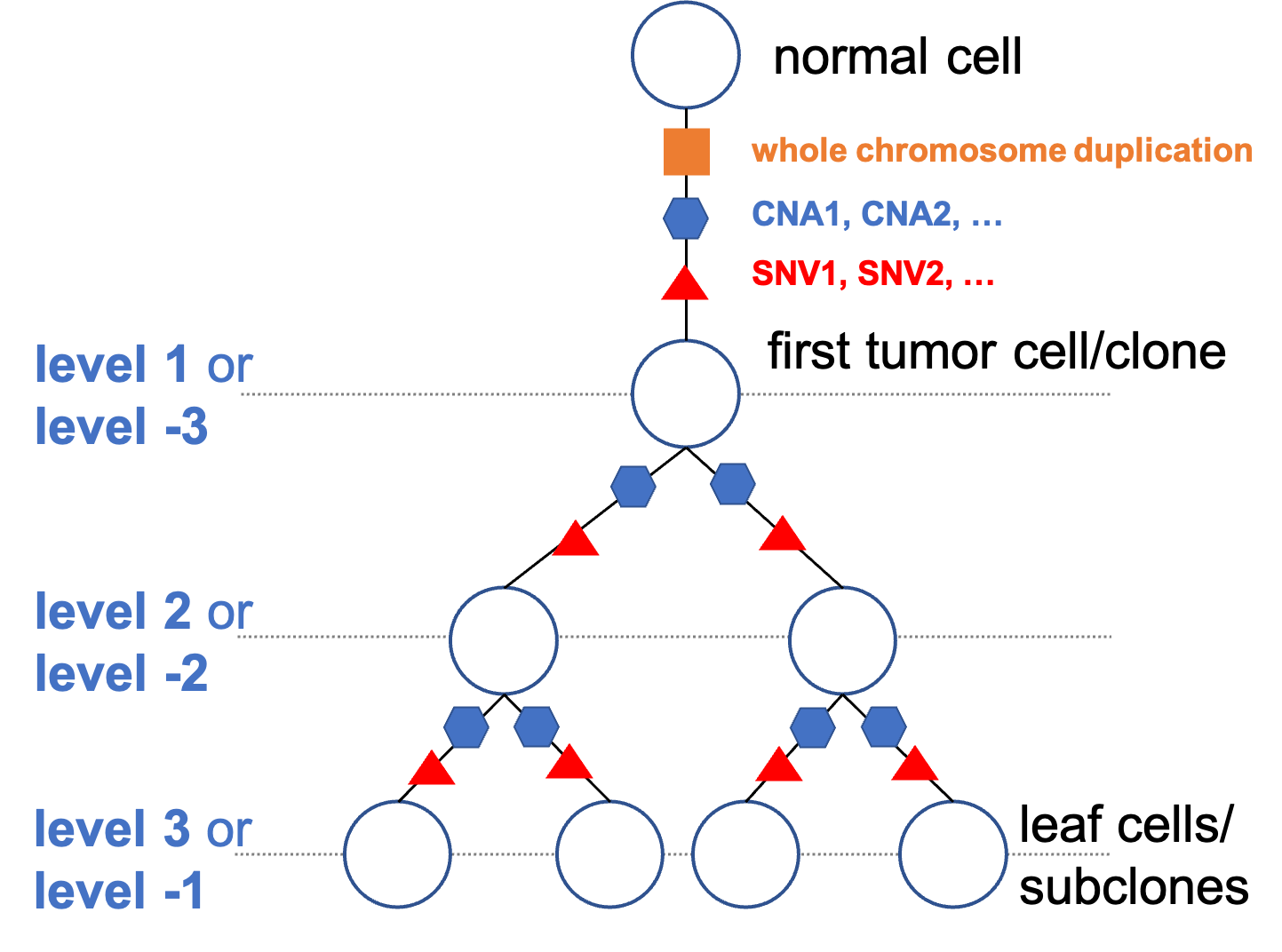
-L (--levels) This is for both tree inference and sampling ancestral nodes. If the user is interested in sequencing nodes on multiple levels, user can specify the levels of interest by separating them by semicolon. The first tumor cell/clone under the trunk has level 1. If counting from the bottom (leaf) of the tree, use minus before the number. For example, -1 is the leaf level. The range of the level should be within [-depth, depth]. Users can specify desired levels according to -G to know which levels are available. If that is the case, use a very small -K to make sure the depth is not smaller than the biggest level you specify. (default: -1)-U (--bulk-levels) The levels of the bulk sequencing separated by semicolon. The definition of the levels is the same as in -L. The default for this option is NA, meaning no bulk sequencing.The following illustration shows how the levels are counted.
-
Parameters controlling bulk sequencing: -V. -V is similar to -v but it applies to the bulk sequencing. Since bulk sequencing is usually of a much deep depth than single cell, use -V >= 5 if bulk sequencing data is desired. The same coverage applies to all ancestral genomes.
-V (--cov-bulk) The coverage of the bulk sequencing. The same for all levels. This parameter is needed when -U is identified. (default: 30) -
Parameters for parallel job submissions: -p and -Y. SimSCSnTree allows parallel processing in simulating reads. Use -p to specify the number of processors. To further parallelize sequencing reads, use -Y to specify a range of nodes to sequence for a job being submitted. Users can specify different ranges of nodes for sequencing for different jobs and thus finish the sequencing in a timely manner. The option -Y shall be in the format of a.b, in which a and b are the smallest and largest numbers of a node on a certain level that the job will process. The node index is 0-based. When -Y is default which is -1, all nodes for the specified level will be sequenced in this job, in which case there is no parallelization for multiple jobs to speed up the sequencing.
-p (--processors) Numbers of processors available.-Y (--leaf-index-range) For parallele job submission. >= min, < max leaf index will be processed. min.max. This counts leaf nodes from 0. (default: -1) -
Parameters for clonality study: -M. When users are interested in the case when each leaf node represents a cell instead of a subclone, -M shall be on. Specify it to be 1 to turn it on.
-M (--single-cell-per-node) If this is on, each node represents one cell and there is no clonality in the node. In this case tree_width will be the same as n (leaf num). 1 is on. (default: 0)
For a complete list of options, type
python main.par.overlapping.py --help
python main.par.overlapping.py -r data --treewidth 8 --treewidthsigma 0.001 --treedepth 4 --treedepthsigma 0.001 --template-ref ~/references/hg19/hg19.fa -m 2000000 -e 5000000 -R 2 -M 1
This command simulates a tree that has about 8 leaf cells (--treewidth 8 --treewidthsigma 0.001) with tree depth 4 (--treedepth 4) with both CNVs and SNVs imputed on the branches. The SNV rate is set up to be 2 (-R 2), and the CNV size is set up to follow an exponential destribution with p=5Mbp (-e 5000000) plus a minimum size of 2Mbp (-m 2000000). The SNV number on a branch will be SNV rate * branch length, whereas branch length is sampled from an exponential distribution with p = 1. Both alleles of the root node start from the hg19 reference file (--template-ref ~/references/hg19/hg19.fa). All .npy files will be stored in data folder (-r data) under the current directory. Remove "data" folder (or back it up to a different name) before running this command to avoid the error message.
python main.par.overlapping.py -k 1 -r data -S ~/github/SimSCSnTree/wgsim-master/ --Lorenz-y 0.28 --template-ref ~/references/hg19/hg19.fa -M 1 -L -1 -Y 0.1 -v 0.01 -l 70
This command runs step 2 to sample reads (-k 1). It reads the .npy files from data folder (-r data), runs wgsim in ~/github/SimSCSnTree/wgsim-master/ (-S ~/github/SimSCSnTree/wgsim-master/) to simulate reads. Notice that the reference file needs to be specified (--template-ref ~/references/hg19/hg19.fa) as the .npy files from the first step does not store any fasta file for the sake of space. The reads are simulated only from the leaf level (-L -1) and each node at the leaf level represents only one cell (-M 1). Given -Y 0.1, this command simulates only the first leaf cell. 0 represents the start of the index of the cell of interest, and 1 is the end of the index of the cell of interest. Both start and end are zero-based. The cell at the end, 1 in this case, is not included in the sequence. If the first three cells are to be sequenced, specify with -Y 0.3. Notice --Lorenz-y is set to be 0.28, which is correponding to the read depth fluctuation from DOP-PCR. For reference, when --Lorenz-x is fixed to the default value (0.5), 0.38 corresponds to the bulk sequencing, and 0.27 corresponds to sequencing from MALBAC. The average read coverage is 0.01X (-v 0.01) and the read length is 70bp for each end for paired-end sequencing. -p 8 means there are eight processors to be used to finish this task and SimSCSnTree implemented the parallelization to use all processors specified in the sequencing step.
python main.par.overlapping.py -k 1 -r data -S ~/github/SimSCSnTree/wgsim-master/ --Lorenz-y 0.28 --template-ref ~/references/hg19/hg19.fa -M 1 -L "1;2;3" -Y 0.1 -v 0.01 -l 70
The difference between this command and the previous one is that instead of sequencing the leaf level (-L -1), it sequences at the level of 1, 2 and 3 whereas 1 corresponds to the first tumor cell under the trunk that connects this tumor cell and a normal cell. Here -Y 0.1 means only the first cell for every specified level will be sequenced.
python main.par.overlapping.py -k 1 -r data -S ~/github/SimSCSnTree/wgsim-master/ --Lorenz-y 0.28 --template-ref ~/references/hg19/hg19.fa -n 100 -L -1 -Y 0.1 -v 0.001 -l 70
This command does not specify that a node is a single cell (no -M 1) and thus refers to a clonality study in which each node corresponds to multiple cells. The distribution of the cells is according to the Beta splitting model of the tree from 100 cells in total on the leaf (-n 100). Notice here we run on only the first leaf node using -L -1 -Y 0.1, and thus the number of cells being sequenced is not necessarily 100, but 100 multiplied by the percentage of the first leaf node on the leaf level. If -Y is not specified, then the summation of all cells being sequenced on the leaf level will be 100. If -L is "1;2;3" and no -Y is specified, then at each specified level (1, 2 and 3), 100 cells will be sequenced.
python main.par.overlapping.py -k 1 -r data -S ~/github/SimSCSnTree/wgsim-master/ --Lorenz-y 0.28 --template-ref ~/references/hg19/hg19.fa -M 1 -L -1 -Y 0.1 -v 0.001 -U -1 -V 5 -l 70
This command, in addition to sequencing the cell on the first leaf node, sequences also the bulk sample on the leaf level (-U -1) at the coverage of 5X (-V 5). SimSCSnTree sequences bulk sample before single cells if both are desired.
The following examples have also appeared in the previous version of SimSCSnTree but edited according to the current new version. The previous version of SimSCSnTree can be found in "Assessing the performance of methods for copy number aberration detection from single-cell DNA sequencing data" authored by XFM, ME, NN and LN in 2020.
python main.par.overlapping.py -r data_large -S ~/github/SimSCSnTree/wgsim-master/ --template-ref ~/references/hg19/hg19.fa --treewidth 50 --treewidthsigma 0.001 -X 8 -W 1 -C 0.3 -E 1 -J 1 -m 2000000 -e 5000000
This command simulates a large tree with 50 leaf nodes (--treewidth 50 --treewidthsigma 0.001). On the trunk, there are eight times more CNAs on the trunk than the other branches on the tree (-X 8), and there is whole chromosome amplification on the trunk (-W 1), whereas for each chromosome, the possibility that it has the amplification is 30% of chance (-C 0.3). If a chromosome is selected to have amplification on the trunk, the number of copies to be amplified is 1 (from -E 1) multiplied by a random draw from a geometric distribution whose p is 1 (-J 1). A CNA's size is 2000000 (-m 20000000) plus a random draw from an exponential distribution whose p is 5000000 (-e 50000000).
The following lists the command to simulate the tree and the alternative alleles (step 1 of the simulator) for different ploidies.
-
Ploidy 1.55
python main.par.overlapping.py -r data_ploidy_1p55 -S ~/github/SimSCSnTree/wgsim-master/ --template-ref ~/references/hg19/hg19.fa --treewidth 8 --treewidthsigma 0.001 -X 25 -W 0 -m 2000000 -e 5000000 -d 1 -c 3
This command simulates a tree with eight leaf nodes (--treewidth 8 --treewidthsigma 0.001) and has twenty-five times more CNAs on the trunk than the other branches (-X 25). The number of CNAs on the other branches is a random draw from a poission distribution whose lambda is 3 (-c 3). There is no whole chromosome amplification (-W 0). CNAs' sizes are 2000000 (-m 20000000) plus a random draw from an exponential distribution whose p is 5000000 (-e 50000000), and all of the CNAs are copy number loss (-d 1). The resulting npy files will be stored in newly made folder data_ploidy_1p55 (-r data_ploidy_1p55).
-
Ploidy 2.1
python main.par.overlapping.py -r data_ploidy_2p1 -S ~/github/SimSCSnTree/wgsim-master/ --template-ref ~/references/hg19/hg19.fa --treewidth 8 --treewidthsigma 0.001 -X 8 -W 1 -C 0.05 -m 2000000 -e 5000000
This command simulates a tree with eight leaf nodes (--treewidth 8 --treewidthsigma 0.001) and has eight times more CNAs on the trunk than the other branches (-X 8). There is whole chromosome amplification (-W 1) and the possibility that a chromosome is amplified is 5% of the chance (-C 0.05). CNAs' sizes are 2000000 (-m 20000000) plus a random draw from an exponential distribution whose p is 5000000 (-e 50000000). The resulting npy files will be stored in newly made folder data_ploidy_1p55 (-r data_ploidy_2p1).
-
Ploidy 3.0
python main.par.overlapping.py -r data_ploidy_3 -S ~/github/SimSCSnTree/wgsim-master/ --template-ref ~/references/hg19/hg19.fa --treewidth 8 --treewidthsigma 0.001 -X 8 -W 1 -C 0.5 -m 2000000 -e 5000000
The major difference between this command and the previous one for ploidy=2.1 is that there is now 50% of the chance for a chromosome to have whole chromosome amplification on the trunk (-C 0.5) instead of 5%.
-
Ploidy 3.8
python main.par.overlapping.py -r data_ploidy_3p8 -S ~/github/SimSCSnTree/wgsim-master/ --template-ref ~/references/hg19/hg19.fa --treewidth 8 --treewidthsigma 0.001 -X 8 -W 1 -C 0.9 -m 2000000 -e 5000000
The major difference between this command and the previous one for ploidy=3.0 is that there is now 90% of the chance for a chromosome to have whole chromosome amplification on the trunk (-C 0.9) instead of 50%.
-
Ploidy 5.26
python main.par.overlapping.py -r data_ploidy_5p26 -S ~/github/SimSCSnTree/wgsim-master/ --template-ref ~/references/hg19/hg19.fa --treewidth 8 --treewidthsigma 0.001 -X 8 -W 1 -C 0.9 -J 0.55 -m 10000000 -e 5000000
Two major differences between this command and the previous one for ploidy=3.8 are 1) the number of whole chromosome amplification copy equals to a multiplier, which is 1 since -E 1 by default, multiplied by a random draw from a geometric distribution whose p equals to 0.55 (-J 0.55) instead of 1; 2) the minimum CNA size is 10000000 (-m 10000000) instead of 2000000.
The following lists the command to simulate the reads (step 2 of the simulator) for different fluctuations. Notice the major difference is on -y.
-
MALBAC
python main.par.overlapping.py -r data_fluctuation_MALBAC -S ~/github/SimSCSnTree/wgsim-master/ --template-ref ~/references/hg19/hg19.fa -l 36 -x 0.5 -y 0.27 -k 1 -
DOP-PCR
python main.par.overlapping.py -r data_fluctuation_DOPPCR -S ~/github/SimSCSnTree/wgsim-master/ --template-ref ~/references/hg19/hg19.fa -l 36 -x 0.5 -y 0.28 -k 1 -
TnBC
python main.par.overlapping.py -r data_fluctuation_TnBC -S ~/github/SimSCSnTree/wgsim-master/ --template-ref ~/references/hg19/hg19.fa -l 36 -x 0.5 -y 0.33 -k 1 -
Bulk
python main.par.overlapping.py -r data_fluctuation_bulk -S ~/github/SimSCSnTree/wgsim-master/ --template-ref ~/references/hg19/hg19.fa -l 36 -x 0.5 -y 0.38 -k 1
-
Read the from_first_step.tree.npy file in the folder specified by -r option generated in the first step of the simulator and convert it to a csv file.
python read_tree.py -s -f from_first_step.tree.npy > gt.all.csvThis step generates a file in bed format that contains all ground truth CNAs for each cell. The fourth column (1-based) is the cell ID.
-
Generate the ground truth for each individual cell from gt.all.csv.
mkdir gt_seppython comparison/sep_groundtruth.py gt.all.csv gt_sep/gtThis script will generate ground truth file for each cell in gt_sep folder, with the prefix gt.
-
If you want to generate segcopy formatted ground truth file for comparison, use the following command.
python comparison/bin_groundtruth.py -a segcopy_f -b gt.all.csv --leafonly(optional) > gt.all.segcopyformattedsegcopy_f is a file you generated from Ginkgo named SegCopy. We use this file as a template to generate a file with the same format. gt.all.csv is the file you generated in step #1 in this section. If you want to include all ancestral nodes along with the leaves, do not put the option --leafonly in the command line. Otherwise use --leafonly in your command.
The format of the output is "chr, start, end, leaf#1, leaf#2, ...". From the fourth column (1-based), the entries are integer copy numbers.
Read the from_first_step.tree.npy file in the folder specified by -n option generated in the first step of the simulator and convert it to a csv file.
python read_tree.py -n -f from_first_step.tree.npy > snv.all.csv
The resulting file's format is chromosome, position, reference nucleotide, variant nucleotide, the allele the variant is on (0 or 1), and the ID of the cell that contains this SNV.
The simulator automatically stores the tree structure in from_first_step.tree.npy. To generate the newick string of the tree, use the following command.
python gen_newick.py from_first_step.tree.npy > newicktree.txt