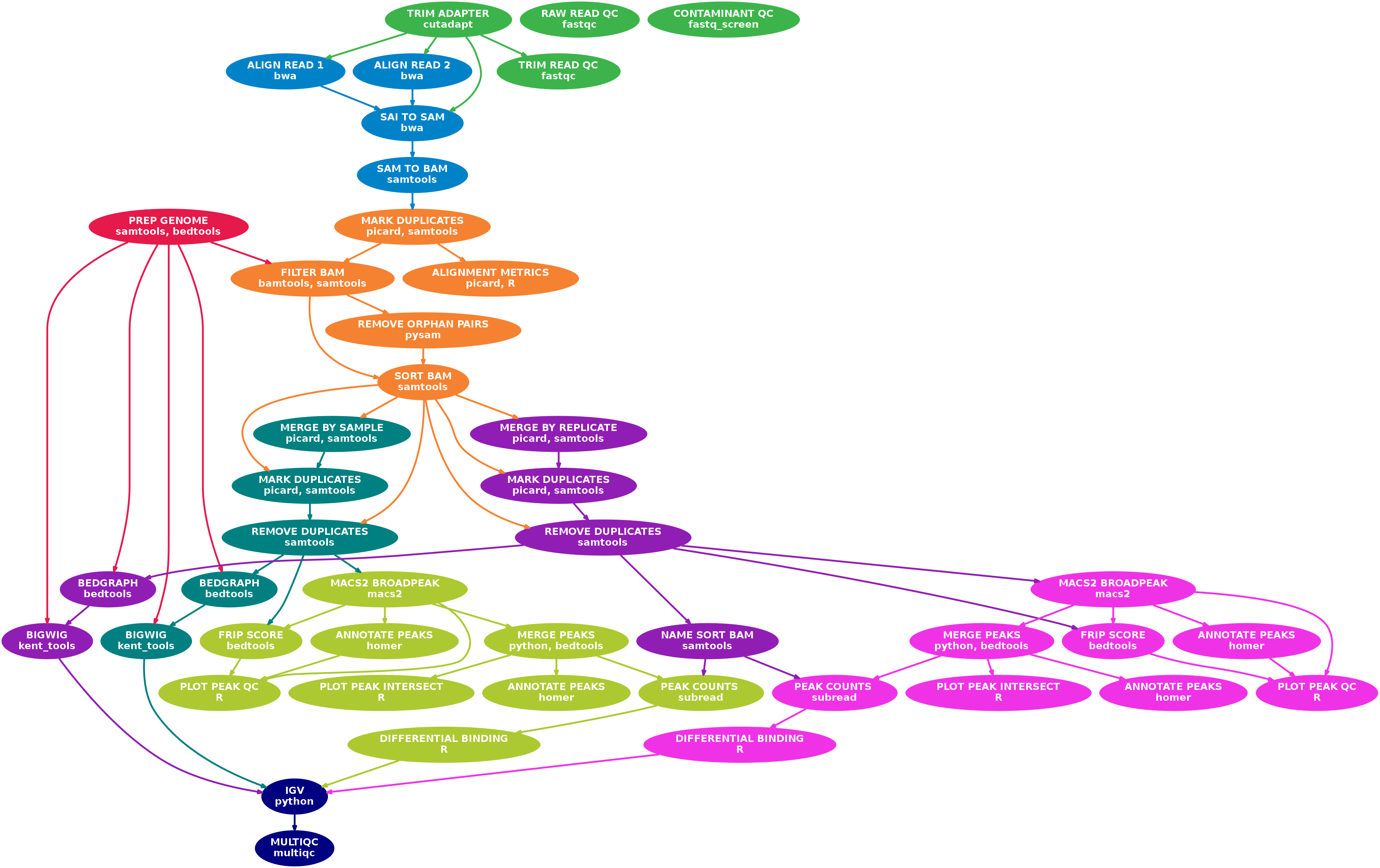
A Nextflow pipeline for processing paired-end Illumina ATACSeq sequencing data.
The pipeline was written by The Bioinformatics & Biostatistics Group at The Francis Crick Institute, London.
- Raw read QC (
FastQC,Fastq Screen) - Adapter trimming (
cutadapt) - Alignment (
BWA) - Mark duplicates (
picard) - Filtering to remove:
- reads mapping to mitochondrial DNA (
SAMtools) - reads mapping to blacklisted regions (
SAMtools,BEDTools) - reads that are marked as duplicates (
SAMtools) - reads that arent marked as primary alignments (
SAMtools) - reads that are unmapped (
SAMtools) - reads that map to multiple locations (
SAMtools) - reads containing > 3 mismatches in either read of the pair (
BAMTools) - reads that have an insert size > 2kb (
BAMTools) - reads that are soft-clipped (
BAMTools) - reads that map to different chromosomes (
Pysam) - reads that arent in FR orientation (
Pysam) - reads where only one read of the pair fails the above criteria (
Pysam)
- reads mapping to mitochondrial DNA (
- Merge alignments at replicate and sample level (
picard)- Re-mark duplicates (
picard) - Remove duplicate reads (
SAMtools) - Create normalised bigWig files scaled to 1 million mapped read pairs (
BEDTools,wigToBigWig) - Call broad peaks (
MACS2) - Annotate peaks relative to gene features (
HOMER) - Merge peaks across all samples and create tabular file to aid in the filtering of the data (
BEDTools) - Count reads in merged peaks from replicate-level alignments (
featureCounts) - Differential binding analysis, PCA and clustering (
R,DESeq2)
- Re-mark duplicates (
- Create IGV session file containing bigWig tracks, peaks and differential sites for data visualisation (
IGV). - Collect and present QC at the raw read, alignment and peak-level (
MultiQC,R)
The documentation for the pipeline can be found in the docs/ directory:
- Installation
- Pipeline configuration
- Reference genome
- Design file
- Running the pipeline
- Output and interpretation of results
- Troubleshooting
The pipeline was written by the The Bioinformatics & Biostatistics Group at The Francis Crick Institute, London.
The pipeline was developed by Harshil Patel, Philip East and Nourdine Bah.
The NGI-RNAseq pipeline developed by Phil Ewels was used a template for this pipeline. Many thanks to Phil and the team at SciLifeLab. The help, tips and tricks provided by Paolo Di Tommaso were also invaluable. Thank you!
This project is licensed under the MIT License - see the LICENSE.md file for details.