Coordinating Tutorial Fixes #2112
Comments
|
I'll start on the two GAN tutorials. The CGAN one is very out of date. I think it would be best to convert it to use the GAN class. |
|
I'm starting on tutorial 2. |
|
I'm looking into tutorial 12 now. I suggest we just get rid of tutorial 20, the one on converting models to estimators. We don't support |
|
I'm getting an error in tutorial 12 that I'm not sure what to do about. When it tries to evaluate the performance on
Looking at the original data file desc_canvas_aug30.csv I can confirm this is true. The code tells it to load the |
|
+1 for getting rid of tutorial 20! Estimators seem to be on the way out in TensorFlow so seems less useful. Good point on tutorial 12. Perhaps we can switch the metric to something like recall on the positive class or FPR/TPR that would be meaningful for a dataset with only positives? (Assuming the datapoints are positives) |
|
Why is the tutorial testing on a dataset with only positives in the first place? That doesn't seem like a very useful test. Can you explain what the crystal dataset is? |
|
That tutorial was based on this paper (https://pubs.acs.org/doi/abs/10.1021/acs.jcim.6b00290). It's been a few years, but I recall that that dataset was testing whether the model trained on assay data could generalize to a separate dataset (compounds which had been co-crystallized with protein/ligand binding). Since these were crystal structures, they were only positives (since we only had binding structures). If this is getting too into the weeds, might be totally fine just to remove from the tutorial. It might be too much into the weeds for beginners and may not be helping build comprehension |
|
What if we just remove the checks against that dataset? We already print results for the train, validation, and test datasets. That seems like plenty for a tutorial. |
|
Sounds good! |
|
I'm reaching the conclusion that notebook is just a mess. I'm trying to figure out what it's doing and why, but with only partial success. Here's what I've figured out so far. It begins by downloading two CSV files and loading them into memory. It confusingly assigns them to variables called It plots a bunch of molecules from both files, but there's no explanation of why it does this or what we're supposed to learn from them other than the one vague sentence, "Note the complex ring structures and polar structures." It now uses a UserCSVLoader to create actual Datasets from those files. Just to be extra confusing, it assigns them to the same variables it previously used for the DataFrames, It splits it into train, validation, and test datasets. It comments without explanation that the test set is really a validation set and the validation set is really a test set. I have no idea what it's talking about. It also applies normalization and clipping transformers. The text says these are being applied to the pIC50 values, which is false: as noted above, the Dataset used the "Class" column, not the "pIC50" column. It fits RandomForestClassifiers and MultitaskClassifiers to the training set (again emphasizing that the labels are classes, not continuous values, so the transformers made no sense) and computes the AUC on the various datasets. As noted above, this produces an exception for the crystal dataset because every sample has the same class. I don't see any way this code could ever have worked. Now it loads the files yet again, only this time loading the "pIC50" column. (And yet again reusing the same variables!) It applies the transformers, fits regression models, and computes R^2 on them. Except that it gets an R^2 of 0 for the crystal dataset, because that file doesn't contain any pIC50 values. The column is completely blank. By the way, it refers to the dataset as "BACE". Is that the same BACE dataset that's already included in molnet? If so, why go through the business of downloading csv files rather than just loading it from molnet? Anyway, as it currently exists this notebook is not a tutorial. It's just a bunch of code, some of it nonfunctional, with very little explanation of what it's doing or why. Perhaps it could be turned into a tutorial, but that would involve a major rewrite. If you can explain what's going on with the data, and what we want the user to learn from reading the tutorial, I could attempt to do that. |
|
I agree, this one is a mess! This is the same BACE that we have in MoleculeNet. I think the original version of this tutorial predates the moleculenet version (hence the archaic loading). Perhaps one way we could cleanly rework it is to just make it a tutorial about working with the BACE dataset from MoleculeNet. The difference between this and the earlier solubility tutorials would be that we would be learning to predict protein-ligand interaction instead of a chemical property, but structurally the tutorial should probably match our earlier tutorial for property prediction on MoleculeNet |
|
I will try to fix tutorial 4 next.
…On Tue, 25 Aug 2020 at 04:55, Bharath Ramsundar ***@***.***> wrote:
I agree, this one is a mess! This is the same BACE that we have in
MoleculeNet. I think the original version of this tutorial predates the
moleculenet version (hence the archaic loading). Perhaps one way we could
cleanly rework it is to just make it a tutorial about working with the BACE
dataset from MoleculeNet.
The difference between this and the earlier solubility tutorials would be
that we would be learning to predict protein-ligand interaction instead of
a chemical property, but structurally the tutorial should probably match
our earlier tutorial for property prediction on MoleculeNet
—
You are receiving this because you were mentioned.
Reply to this email directly, view it on GitHub
<#2112 (comment)>,
or unsubscribe
<https://github.com/notifications/unsubscribe-auth/AAOVM37PBYOCFSYTTUFR6ITSCMRYLANCNFSM4QHXISPQ>
.
|
|
I'll also do tutorial 5 while I'm at it. It seems to have some of the same problems. |
|
I am trying to fix the tutorial 4. It seems like I need to have some understanding of graph conv model and also featurization for the model. I am trying to understand the relevant parts of Is there some more documentation for it? Or some other material that I can read (I am interested in both understanding graph conv model and featurization for it)? If not, I can try to spend more time through Tensorflow, Keras documentation and DeepChem code. |
|
On tutorial 5 I'm running into the same error about, "Only one class present in y_true." This one uses the MUV dataset. Out of 93,000 samples, no task has more than 30 positives, and a lot have even less. And there are 17 tasks. The odds are very high that either the validation set or test set will randomly end up with no positives at all for at least one task, and then we get an error. How do you suggest dealing with this? |
|
@neel-shah Unfortunately there isn't good documentation for the usage of these intermediate arrays. I've written up some docs for |
|
@peastman Perhaps |
|
|
|
Yet another issue I'm running into on tutorial 12. If I tell it to use a random split, the random forest and MultitaskRegressor both do reasonably well: But the documentation for BACE recommends using scaffold splitting instead. (So why isn't that the default? Seems like a bug to me.) And when I do that, the generalization performance is terrible. In fact, the validation set comes out strongly anticorrelated with the model predictions. Any suggestions? |
Thanks @rbharath, the docs helped indeed. I think I now have decent understanding about graph conv to get through tutorial 4 (or at least I hope). Although I am stuck at an error which looks like the code in the tutorial should never have worked in the first place or there are some major changes happened underneath. So at one point in tutorial 4 it talks about creating graph conv model from scratch. For that we first build the layers of the model and we build the model using DeepChem's graph conv model class which throws the following error The error looks legit to me because we are trying to split Do I get it right? Maybe we should instantiate |
|
Just a guess: try changing the definition of membership = layers.Input(shape=(1,), dtype=tf.int32) |
|
Thanks! I still see the same error after changing The error comes from this line. |
|
@neel-shah The issue here is that this tutorial was originally written for TF1.x. Unfortunately, for TF 2.2, we had to change |
|
@peastman Hmm, that's really weird. From a purely scientific viewpoint, scaffold split is better since it tests the ability of the model to generalize to new scaffolds, but it's clear the model doesn't generalize well to new scaffolds. It's really strange to see the anti-correlation. Perhaps one way to handle this in the tutorial would be to make it pedagogical. That is, we can show random split works well, then show scaffold split doesn't. And explain that this means the learned model doesn't actually generalize well to new scaffolds, so users should avoid using it to make predictions on structurally dissimilar molecules. |
|
We discussed this a bit on Friday. I'm posting here to continue the discussion. In addition to fixing broken code, a lot of the tutorials need deeper changes. Many of them weren't really designed to be tutorials. They were just notebooks containing sample code. We collected them together and arranged them in a sequence, but they don't really form a coherent set of lessons. I suggest we overhaul the full set of tutorials from beginning to end. Each one should have a specific lesson it teaches, and they should build on each other in a logical way so a user can learn to use DeepChem by reading through the tutorials in order. We also discussed another tutorial we should add: one that introduces the KerasModel and TorchModel classes, showing users how to use TensorFlow and PyTorch based models with DeepChem. I can write that, if you want. |
|
@peastman A tutorial introducing the KerasModel and TorchModel classes would be a great addition! That would be very welcome :) I definitely agree that we need to overhaul the tutorials to form a coherent set of lessons as you suggest. The tutorial set has grown organically, which means we have a lot of cruft in there that doesn't really contribute. Do you have any thoughts on a potential coherent lesson structure? I can also take a look to see what might make sense |
|
I suggest we make up a list of the tutorials we want to have, and what lessons the reader should learn from each one. For example,
And so on. A lot of our current tutorials cover more advanced and specialized subjects, which is fine. We just need to make sure they come later in the sequence, after the reader has already learned everything they need to understand them. |
|
I finally got the code in tutorial 4 to work, this is what model looks like: And this is the result: To be honest I don't understand a lot of things getting executed under the hood. I will spend some time understanding those things which probably will also help to improve code description. |
|
The Butina Clustering code itself has no way of specifying sizes. So you sort of just pick a similarity value you're happy with and see what comes backout. The code really wasn't meant for production |
|
Would it be reasonable to make it work the same way scaffold splitter does? The two methods are conceptually pretty similar. Once you identify scaffolds or clusters, the code for assembling them into datasets could be mostly identical. |
|
That seems reasonable to me. |
|
Tutorials 13 and 14 both need a lot of changes. 13 is written to download the datafiles and process them by hand, instead of just calling |
|
Yep, absolutely, will take these on! I'm planning to hit tutorial 3 this week and will hit these two in the next couple of weeks. |
|
Here's an updated version of the sequence and the status of each one.
There are a few current tutorials that don't appear anywhere in the sequence as currently outlined: learning embeddings with seq2seq (11), synthetic feasibility scoring (15), introduction to bioinformatics (21). What do we want to do with them? Of the tutorials marked TODO, some need only minor revisions and moving into the correct position in the sequence. Others need bigger changes, and a few are completely new tutorials that need to be written. [EDITED: Added in the other three tutorials. Updating status of tutorials as they get done.] |
Anyone have thoughts on this? |
|
Hmm, good question. Here are a couple thoughts:
|
|
Thanks! I edited the list above to add them at the positions you suggested. |
|
Since I'm working on docking, I'd be happy to take on Tutorial 12: Interaction Fingerprints (from current tutorial 13). This could be an expanded, end-to-end example of taking protein-ligand complexes, featurizing them with the interaction fingerprints from #2212, using |
|
Awesome thanks @ncfrey! |
|
I'm trying to update the tutorial on model interpretability (currently number 8). Things are fine up until cell # We can visualize which fingerprints our model declared toxic for the
# active molecule we are investigating
Chem.MolFromSmiles(list(my_fp[242])[0])That magic number 242 seems to come out of nowhere. It appears in this and the following cells, none of which has any explanation. Many of them don't even produce output. The tutorial then declares we've learned something useful, but if so, I don't know what it is! Any explanation of what's going on? |
|
Ah this completely slipped through my inbox! My understanding of this tutorial is that it really is a magic number. The Honestly, I'm not sure we've actually learned anything here. It's useful to look at the contributing fragments and I think it does help chemists who have a direct intuition from the molecules, but it is more pretty pictures than anything. I'm super open to ways we could change up this tutorial so it is more useful and actually teaches something good. |
|
After more work on the interpretability tutorial, I'm starting to have doubts about whether the approach it uses is useful at all. I get a lot of outputs like this:
That shows that this particular model is strongly predicted to be toxic. But when it breaks the prediction down into signals from particular fragments, all the top ones indicate it should be non-toxic! How can that be? Well, when I scroll down further on the list I do start to get lots of ones that lead to the toxic prediction. But the prediction doesn't come from a small number of fragments that give clear signals. It's a sum of small contributions from hundreds of fragments, with slightly more of those fragments indicating toxic than non-toxic. Furthermore, very few of the contributions are from fragments that are present. Most of the prediction is coming from fragments that aren't present (value of 0.00 along the left side). Perhaps someone more experienced with this tool would know how to get useful insight from it, but I don't. |
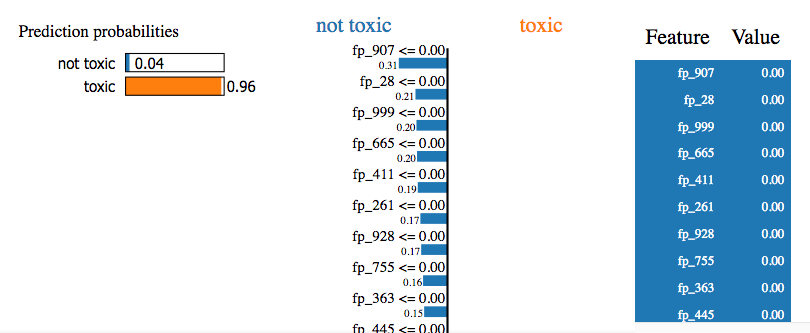
|
CC @lilleswing who has more experience with interpretability and might be able to help out :) I have to confess that I've not had great luck with this interpretabilty technique either, for reasons similar to those you've mentioned. I think sometimes there's a clear signal from one fragment that provides the deciding "vote", but for a lot of cases it seems to be the long tail of many contributions. As a quick question, are the non-present fragments coming from the ECFP hash? That is, it's a sort of hash collision leading the tool to think a fragment with the same hash but not present in the molecule is contributing signal? Perhaps one way we can address this in the tutorial is to just be up-front about these issues and say that "user beware"? |
|
I'm not entirely sure, but I suspect it's more a case of, "This fragment is associated with molecules being toxic. Since it isn't present, that's a strong signal that this molecule is non-toxic." LIME works by building a local linear model, so it can't deal very well with non-linear logic like, "The molecule is toxic if any one of these fragments is present." |
|
I was looking into the bioinformatics tutorial. I'm a bit confused, because it has nothing to do with DeepChem. It's a tutorial on how to do basic sequence manipulation with BioPython. It does end with a cell that says, "TODO(rbharath): Continue filling this up in future PRs." Was this meant to be just the start of a tutorial, and it never got finished? If so, what else was it meant to include? |
|
@peastman Yes you're right! Unfortunately this was to be the start of a sequence and it never got finished. The goal was to work up to the material we present in the book chapters on bioinformatics (or other ML in bioinformatics applications like transcription factor binding site prediction) |
|
Can I take on fixes to README.md of tutorials? I would like to fix the links in the README.md file (some links are outdated or broken). One more thing which I would like to suggest is organizing of tutorial links around functionalities of DeepChem in README.md. We have close to 30 tutorials. Maybe we could organize them into categories like introductory tutorials of DeepChem, tutorials for molecular machine learning, quantum chemistry etc. The end result of README.md will look something like this: /* Introductory content */
Molecular Machine Learning /* Tutorials which are specific to molecular machine learning */
Bioinformatics And so on. |
|
@arunppsg These are all great suggestions! Both the fixing of links and organization of tutorials would add a lot of value. Please go for it :) |
|
I'd be careful about reordering the tutorials. Especially in the earlier ones, each one assumes you've read the ones that come before. |
@arunppsg @rbharath +1 to having a "core functionality" tutorial series that preserves the sequential ordering (maybe 1 - 11?) and introduce all the core features of deepchem. Then splitting into applications as @arunppsg suggested |
|
Tentatively I think we can close this issue for now since we've stabilized the core tutorial series. Will re-open if necessary |
The tutorials need a thorough scrubbing. Let's use this issue to coordinate the needed work and sign up for fixing various tutorials.
CC @peastman @neel-shah
The text was updated successfully, but these errors were encountered: