HMmer Based UndeRstandinG of gene clustERs
A tool to extract and analyse contiguous sets of genes in bacterial genomes, given a concatenated set of protein hidden markov models (HMMs). Written in python 3 and R (for T6SS filtering/grouping)
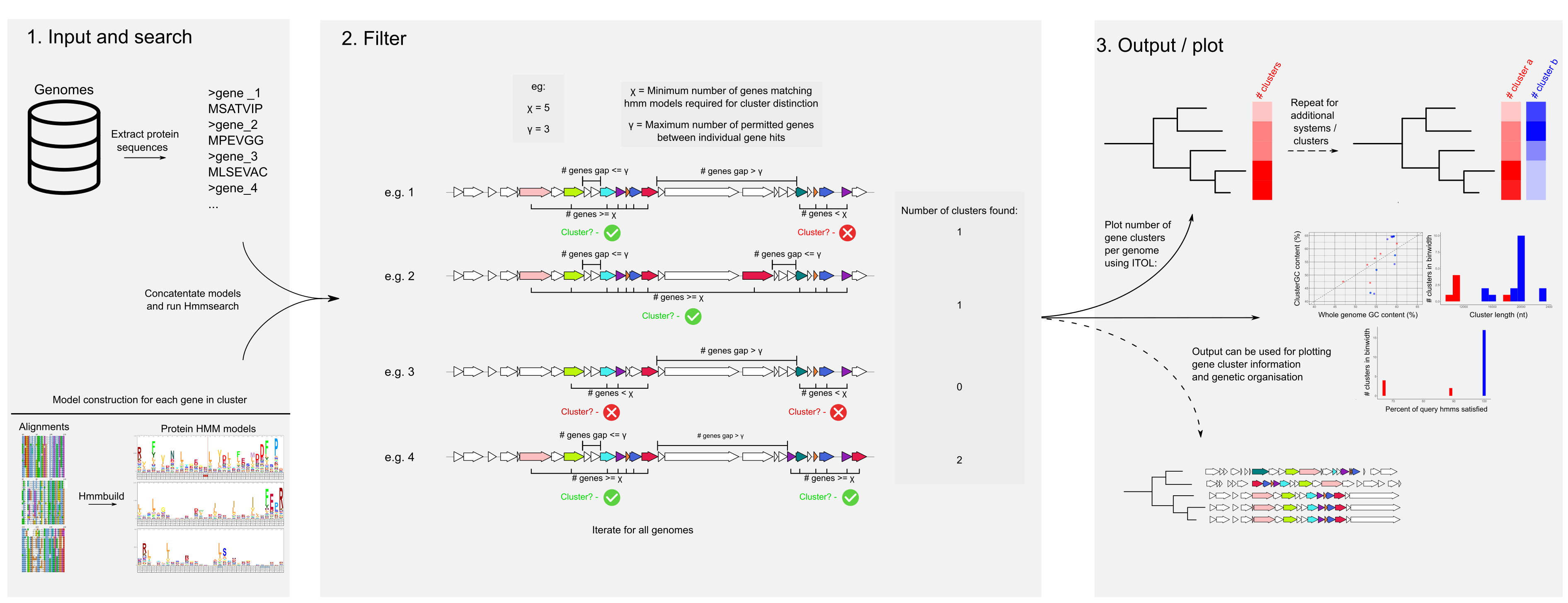
Hamburger uses sets of concatenated HMMs to search for sets of genes co-localised in a genome, using gff3 files as input. Input gff3 files need both the annotation, and fasta sequence (preceded by ##FASTA in the file, as produced by prokka)
Hamburger can be used as follows - where < > denotes user specific addresses/input:
Firstly cloning the git repository using git clone (into desired local directory)
git clone https://www.github.com/djw533/hamburger
Then running hamburger to search using a set of HMMs concatenated into a single file
python <hamburger_directory>/scripts/hamburger.py -g *.gff -i <concatenated set of hmms.hmm> -o <new output directory to write in> -m <minimum number of genes needed to report gene cluster> -l <max permitted gap of non-model genes between genes of interest>
An automated search and subtyping of T6SSs can be used as follows:
python <hamburger_directory>/scripts/hamburger.py -g *.gff -t -o <new output directory to write in>
Followed by a filtering step
python <hamburger_directory>/scripts/post_hamburger_t6SS_search.py -i <output directory used for hamburger search>
Output for loading into itol can be requested with the -q flag
The following dependencies are required:
System:
HMMER
Python 3
(and for T6SS subtyping):
Muscle
Fasttree
R
Python libraries:
Biopython
tqdm
pandas
R libraries (for T6SS subtyping) - can be installed using the install_R_packages.R script:
ggplot2
dplyr
gggenes
RColorBrewer
castor
ape
ggtree
glue
usage: hamburger.py [-h] [-i MANDATORY] [-a ACCESSORY] [-g GFF [GFF ...]] [-f FASTA [FASTA ...]]
[-m MIN_GENES] [-l GENES_GAP] [-u UPSTREAM] [-d DOWNSTREAM] [-c CUTOFF] [-t]
[-n NUM_THREADS] [-k] [-o OUTPUT] [-q] [-w] [-s] [-v]
Extract and plot gene_clusters based on hmm profiles
--------------------HaMBURGER--------------------
-------HMmer Based UndeRstandinG of gene clustERs------
_....----'''----...._
.-' o o o o '-.
/ o o o o o \
__/__o___o_ _ o___ _o _ o_ _ _o_\__
/ \
\___________________________________/
\~`-`.__.`-~`._.~`-`~.-~.__.~`-`/
\ /
`-._______________________.-'
optional arguments:
-h, --help show this help message and exit
-i MANDATORY, --mandatory MANDATORY
Mandatory hmm profile input <required if not using -t flag>
-a ACCESSORY, --accessory ACCESSORY
Accessory hmm profile input
-g GFF [GFF ...], --gff GFF [GFF ...]
Gff file(s) to search. Can be used in combination with --fasta or standalone (if fasta is appended to the end of the gff inbetween a line with ##FASTA)
-f FASTA [FASTA ...], --fasta FASTA [FASTA ...]
Fasta file(s) to search. Can be used in combination with --gff or standalone, in which case prodigal will predict CDSs
-m MIN_GENES, --min_genes MIN_GENES
Minimum number of genes in gene cluster, default = 4
-l GENES_GAP, --genes_gap GENES_GAP
Maximum number of genes gap between hits, default = 10
-u UPSTREAM, --upstream UPSTREAM
Number of nucleotides to include upstream/"right" of gene cluster, default = 0
-d DOWNSTREAM, --downstream DOWNSTREAM
Number of nucleotides to include downstream/"left" of gene cluster, default = 0
-c CUTOFF, --cutoff CUTOFF
Cutoff HMMER score for each hit, default = 20
-t, --t6ss Automatic searching for T6SSs, uses min_genes = 8, genes_gap = 12, mandatory hmm profile of all 13 tss genes
-n NUM_THREADS, --num_threads NUM_THREADS
Number of threads to use, default = 1
-k, --keep_files Keep all intermediate files produced, default = False
-o OUTPUT, --output OUTPUT
Output directory, default =Hamburger output. Will not write over a previously existing output folder!
-q, --itol Create itol output for number of T6SSs and subtypes per strain
-w, --overwrite Overwrite existing blast database directory
-s, --save_gffs Save output gff files
-v, --version Print version and exit