{kind=link}
{kind=link}
Repository containing scripts to perform near-real time tracking of SARS-CoV-2 in Connecticut using genomic data. This pipeline is used by the Grubaugh Lab at the Yale School of Public Health, and results are shared on COVIDTrackerCT.
This repository contains scripts for running pre-analyses to prepare sequence and metadata files for running augur and auspice, and for running the nextstrain pipeline itself.
To be able to run the pipeline determined by the Snakefile, one needs to set up an extended conda nextstrain environment, which will deploy all dependencies (modules and packages) required by the python scripts located at the scripts directory. Check each individual script in that directory to know what they do along the workflow.
Follow the steps below to set up a conda environment for running the pipeline.
Access a directory or choice in your local machine:
cd 'your/directory/of/choice'
Clone this repository ncov
git clone https://github.com/andersonbrito/ncov.git
Rename the directory ncov as you wish. Access the newly generated directory in your local machine, change directory to config, and update your existing nextstrain environment as shown below:
cd 'your/directory/of/choice/ncov/config'
conda env update --file nextstrain.yaml
This command will install all necessary dependencies to run the pipeline, which can be activated by running:
conda activate nextstrain
This minimal set of files and directories are expected in the working directory.
ncov/
│
├── config/
│ ├── auspice_config.json → Auspice configuration file
│ ├── cache_coordinates.tsv → TSV file with pre-existing latitudes and longitudes
│ ├── clades.tsv → TSV file with clade-defining mutations
│ ├── colour_grid.html → HTML file with HEX color matrices
│ ├── dropped_strains.txt → List of genome names to be dropped during the run
│ ├── geoscheme.tsv → Geographic scheme to aggregate locations
│ ├── keep.txt → List of GISAID genomes to be added in the analysis
│ ├── nextstrain.yaml → File used to install nextstrain dependencies
│ ├── reference.gb → GenBank file containing the reference genome annotation
│ └── remove.txt → List of GISAID genomes to be removed prior to the run
│
├── pre-analyses/
│ ├── gisaid_hcov-19.fasta → File with genomes from GISAID (a JSON file can also be used)
│ ├── new_genomes.fasta → FASTA file with the lab's newly sequenced genomes
│ ├── metadata_nextstrain.tsv → nextstrain metadata file, downloaded from GISAID
│ ├── COVID-19_sequencing.xlsx → Custom lab metadata file
│ └── extra_metadata.xlsx → Extra metadata file (can be left blank)
│
├── scripts/ → Set of scripts included in the pipeline
│
├── README.md → Instruction about the pipeline
├── workflow.svg → Diagram showing the pipeline steps
└── Snakefile → Snakemake workflow
Files in the pre-analyses directory need to be downloaded from distinct sources, as shown below.
| File | Source |
|---|---|
| gisaid_hcov-19.fasta provision.json |
FASTA or JSON file downloaded from GISAID |
| new_genomes.fasta¹ | Newly sequenced genomes, with headers formatted as ">Yale-XXX", downloaded from the Lab's Dropbox |
| metadata_nextstrain.tsv² | File 'metadata.tsv' available on GISAID |
| GLab_SC2_sequencing_data.xlsx³ | Metadata spreadsheet downloaded from an internal Google Sheet |
| extra_metadata.xlsx⁴ | Metadata spreadsheet (XLSX) with extra rows, where column names match the main sheet |
Notes:
¹ FASTA file containing all genomes sequenced by the lab, including newly sequenced genomes
² The user will need credentials (login/password) to access and download this file from GISAID
³/⁴ These Excel spreadsheet must have the following columns, named as shown below:
- Filter → include tags used to filter genomes, as set in
rule filter_metadata - Sample-ID → lab samples unique identifier, as described below
- Collection-date
- Country
- Division (state) → state name
- Location (county) → city, town or any other local name
- Source → lab source of the viral samples
The files provision.json and gisaid_hcov-19.fasta contain genomic sequences provided by GISAID. This pipeline takes any of these two file formats as input. provision.json can be downloaded via an API provided by GISAID, which require special credentials (username and password, which can be requested via an application process). Contact GISAID Support <service@gisaid.org> for more information.
The file gisaid_hcov-19.fasta can be generated via searches on gisaid.org. To do so, the user needs to provide a list of gisaid accession numbers, as follows:
- The table below illustrates a scheme to sample around 600 genomes of viruses belonging to lineages
B.1.1.7(alpha variant) andB.1.617.2(delta variant), circulating in the US and the United Kingdom, between 2020-12-01 and 2021-06-30, having other US and European samples as contextual genomes. Note that contextual genomes are selected from two time periods, and in different proportions: 50 genomes up to late November 2020, and 100 from December 2020 onwards. Also, the scheme is set up to ignore genomes from California and Scotland, which means that genomes from those locations will not be included in any instance (they are filtered out prior to the genome selection step). To reproduce the scheme above, the following script can be used, having a--metadatafile listing genomes from GISAID that match those filtering categories:
genome_selector.py [-h] --metadata METADATA [--keep KEEP] [--remove REMOVE] --scheme SCHEME [--report REPORT]
... where --scheme is a TSV file like the one below:
| purpose | filter | value | filter2 | value2 | sample_size | start | end |
|---|---|---|---|---|---|---|---|
| focus | pango_lineage | B.1.1.7 | country | USA | 200 | 2020-12-01 | 2021-06-30 |
| focus | pango_lineage | B.1.617.2 | country | United Kingdom | 200 | 2020-12-01 | 2021-06-30 |
| context | country | USA | 50 | 2020-11-30 | |||
| context | country | USA | 100 | 2020-12-01 | |||
| context | region | Europe | 100 | 2020-12-01 | 2021-06-30 | ||
| ignore | division | California | |||||
| ignore | division | Scotland |
Among the outputs of genome_selector.py users will find text files containing a list of around 550 genome names (e.g. USA/CT-CDC-LC0062417/2021) and a list of gisaid accession numbers (e.g. EPI_ISL_2399048). The first file (with genome names) can be placed in config/keep.txt, to list the genomes that will be included in the build. If a file provision.json is available, the user can use that as the input sequence file, and that list will be used to retrieve the selected genomes. When the JSON file is not available, the second file (with accession numbers) can be used to filter genomes directly from gisaid.org, as follows:
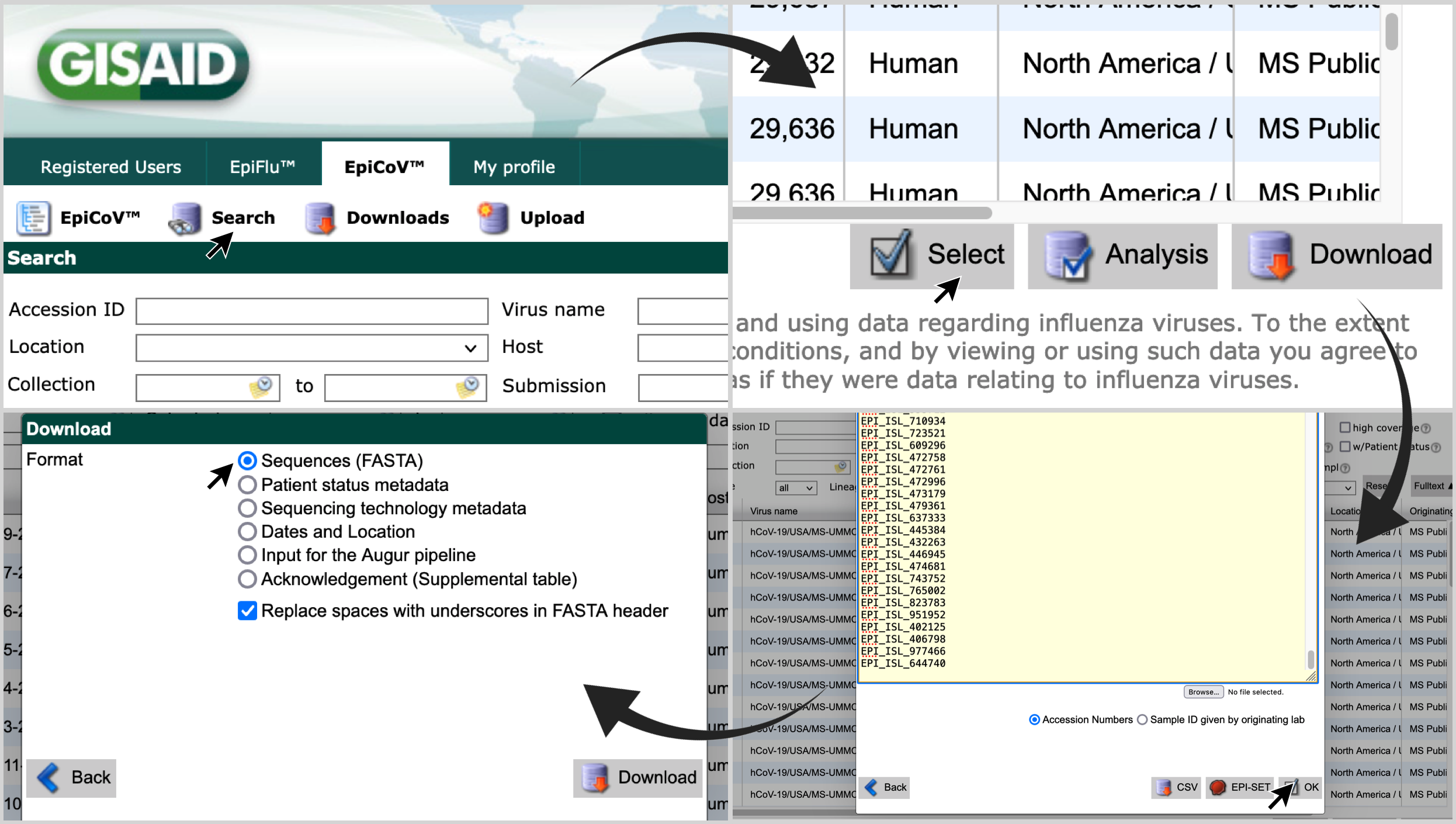
- Access gisaid.org. Login with your credentials, and click on
Search, andEpiCoV™; - Click on
Select, paste the list of accession numbers and click onOK; - Select
Sequences (FASTA)and click onDownload.
The file downloaded via this search can be directly used as the input pre-analyses/gisaid_hcov-19.fasta
By running the command below, the appropriate files sequences.fasta and metadata.tsv will be created inside the data directory, and the TSV files colors.tsv and latlongs.tsv will be created inside the config directory:
snakemake preanalyses
By running the command below, the rest of the pipeline will be executed:
snakemake export
By running the command below files related to previous analyses in the working directory will be removed:
snakemake clean
Such command will remove the following files and directories:
results
auspice
data
config/colors.tsv
config/latlongs.tsv
This command will delete the directory pre-analyses and its large files:
snakemake delete
The code in scripts will be updated as needed. Re-download this repository (git clone...) whenever a new analysis has to be done, to ensure the latest scripts are being used.