{kind=link}
{kind=link}
![]()
Gregory Way and Casey Greene 2018
University of Pennsylvania
The repository stores data and data processing modules to sequentially compress gene expression data.
Named after the mechanical device developed by Alan Turing and other cryptologists in World War II to decipher secret messages sent by Enigma machines, BioBombe represents an approach used to decipher hidden messages embedded in gene expression data. We use the BioBombe approach to study different biological representations learned across compression algorithms and various latent dimensionalities.
In this repository, we compress three different gene expression data sets (TCGA, GTEx, and TARGET) across 28 different latent dimensions (k) using five different algorithms (PCA, ICA, NMF, DAE, and VAE). We evaluate each algorithm and dimension using a variety of metrics. Our goal is to construct reproducible gene expression signatures with unsupervised learning.
Links to access data and archived results can be found here: https://greenelab.github.io/BioBombe/
Compressing gene expression data using multiple latent space dimensionalities learns complementary biological representations Way, G.P., Zietz, M., Rubinetti, V., Himmelstein, D.S., Greene, C.S. Genome Biology (2020) doi:10.1186/s13059-020-02021-3
Our approach is outlined below:
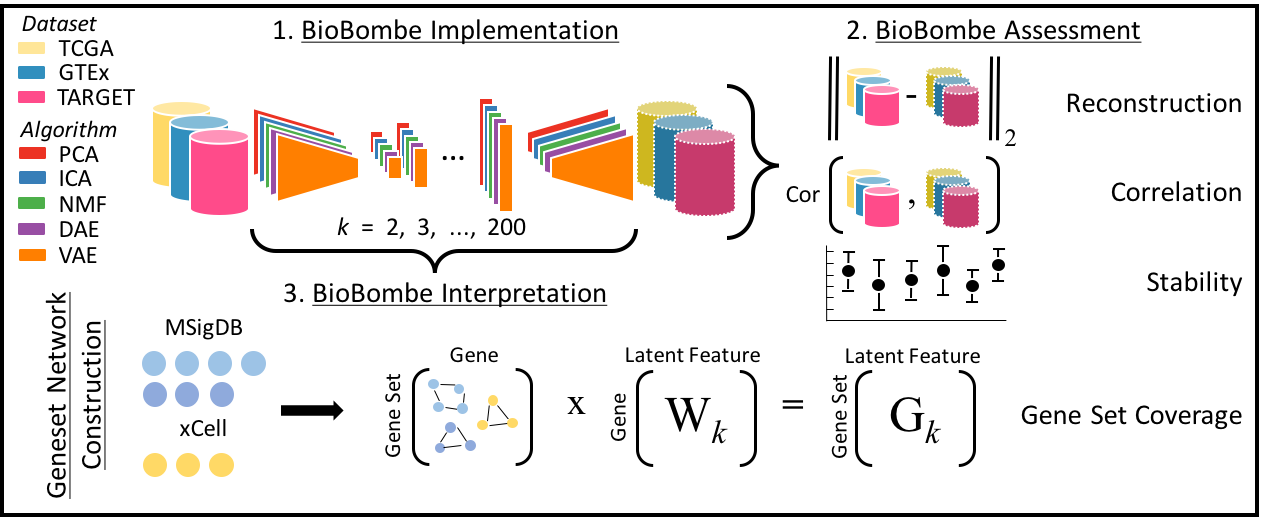
Our model implementation is described below.
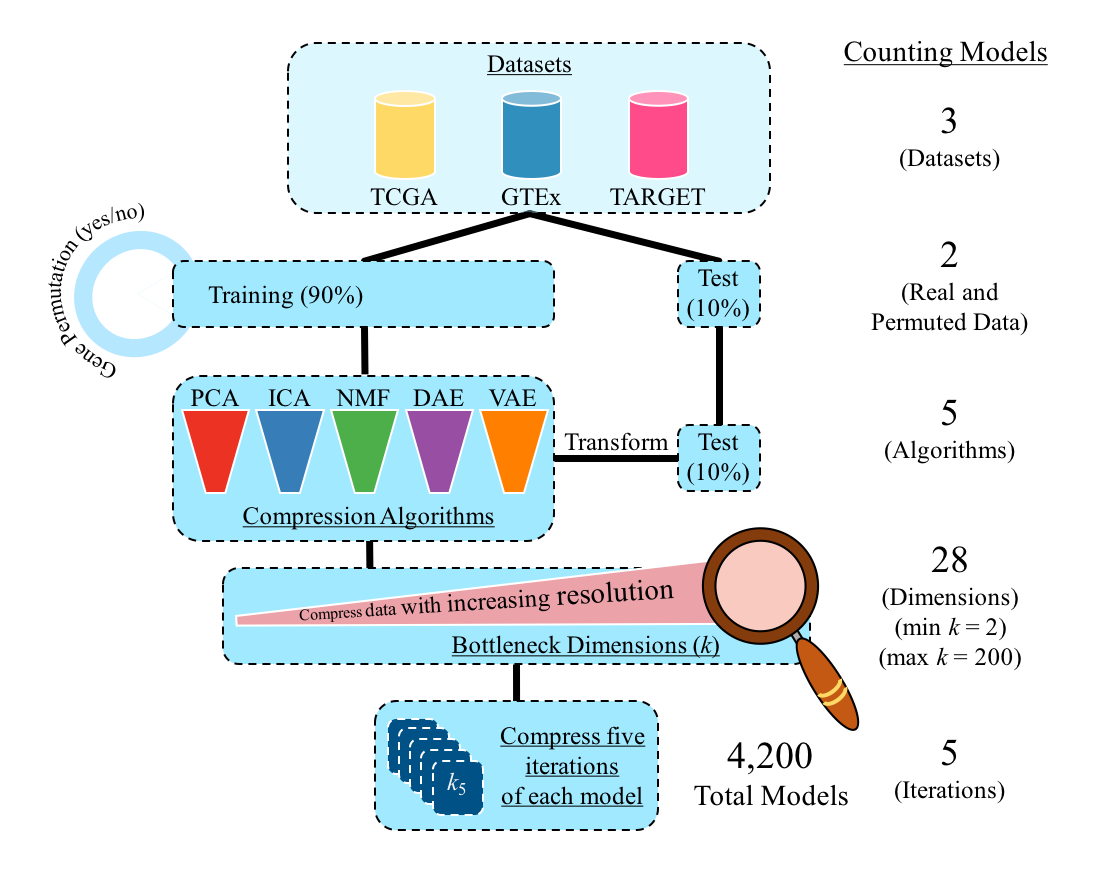
To reproduce the results and figures of the analysis, the modules should be run in order.
| Name | Description |
|---|---|
| 0.expression-download | Download and process gene expression data to run through pipeline |
| 1.initial-k-sweep | Determine a set of optimal hyperparameters for Tybalt and ADAGE models across a representative range of k dimensions |
| 2.sequential-compression | Train various algorithms to compress gene expression data across a large range of k dimensions |
| 3.build-hetnets | Download, process, and integrate various curated gene sets into a single heterogeneous network |
| 4.analyze-components | Visualize the reconstruction and sample correlation results of the sequential compression analysis |
| 5.analyze-stability | Determine how stable compression solutions are between and across algorithms, and across dimensions |
| 6.biobombe-projection | Apply BioBombe matrix interpretation analysis and overrepresentation analyses to assign biological knowledge to compression features |
| 7.analyze-coverage | Determine the coverage, or proportion, of enriched gene sets in compressed latent space features for all models and ensembles of models |
| 8.gtex-interpret | Interpret compressed features in the GTEX data |
| 9.tcga-classify | Input compressed features from TCGA data into supervised machine learning classifiers to detect pathway aberration |
| 10.gene-expression-signatures | Identify gene expression signatures for sample sex in GTEx and TCGA data, and MYCN amplification in TARGET data |
See 2.sequential-compression for more details.
All processing and analysis scripts were performed using the conda environment specified in environment.yml.
To build and activate this environment run:
# conda version 4.5.0
conda env create --force --file environment.yml
conda activate biobombe