Toolkit manual
To improve compatibility, MineProt scripts are coded in native Python3 as much as possible, but they still depend on following third-party packages:
numpy
requests
biopython
Please run pip3 install -r toolkit/scripts/requirements.txt to install them.
Operations will be more convenient with a Bash shell, as several function-integrated shell scripts are provided.
Preprocess your AlphaFold predictions for curation: pick out top-1 model and
generate .cif for visualization.
optional arguments:
-h, --help show this help message and exit
-i I Path to input folder. THIS ARGUMENT IS MANDATORY.
-o O Path to output folder. THIS ARGUMENT IS MANDATORY.
-n N Naming mode: 0: Use prefix; 1: Use name in .a3m; 2: Auto rename;
3: Customize name.
--url URL URL of PDB2CIF API.
-
Make temporary folder
MP-Temp-XXXX; -
Scan the
-idirectory and locate result folders;AlphaFold will generate one subdirectory for one prediction in its
output_dir. Please make sure your-imatches your AlphaFold's--output_dir. -
For each result folder:
- Copy
ranked_0.pdbtoMP-Temp-XXXX; - Add text #Added by MineProt toolkit to the top of
msas/bfd_uniref_hits.a3m(ormsas/bfd_uniclust_hits.a3mfor old versions) and modify its sequence identifiers to UniProt protein accessions before copying it toMP-Temp-XXXX; - Read the
ranking_debug.jsonand locate the PKL file for the best model; export vital model scores from it into a JSON file inMP-Temp-XXXX;
- Copy
-
Rename all files in
MP-Temp-XXXXaccording to-nparameters and move them to the-odirectory; -
Generate CIF files using MineProt PDB2CIF API.
The -o directory will have the following structure:
<output_dir>/
<protein1>.a3m <protein1>.json <protein1>.pdb <protein1>.cif
<protein2>.a3m <protein2>.json <protein2>.pdb <protein2>.cif
...
For each protein, contents of output file are as follows:
-
.a3m- MSA file generated by AI system -
.json- Model scores{ "max_pae": maximum predicted aligned error "pae": [[predicted aligned error],[...],...] "plddt": predicted lDDT-Cα "ptm": pTM score } -
.pdb- Structure file generated by AI system -
.cif- Structure file generated by MineProt PDB2CIF API, supporting Mol* visualization in the style of AlphaFold DB
Preprocess your ColabFold predictions for curation: pick out top-1 model and
generate .cif for visualization.
optional arguments:
-h, --help show this help message and exit
-i I Path to input folder. THIS ARGUMENT IS MANDATORY.
-o O Path to output folder. THIS ARGUMENT IS MANDATORY.
-n N Naming mode: 0: Use prefix; 1: Use name in .a3m; 2: Auto rename;
3: Customize name.
-z Unzip results.
-r Use relaxed results.
--url URL URL of PDB2CIF API.
-n 1is especially useful when you you employcolabfold_searchandcolabfold_batchgenerate large scale structure predictions, forcolabfold_searchwill name output A3M files as<order>.a3mby default.
-
Make temporary folder
MP-Temp-XXXX; -
Scan the
-idirectory, locate target A3M, PDB and JSON files and copy them toMP-Temp-XXXX;Please make sure your
-zand-rmatches your ColabFold's--zipand--relax. -
Rename all files in
MP-Temp-XXXXaccording to-nparameters and move them to the-odirectory; -
Generate CIF files using MineProt PDB2CIF API.
The same as outputs of alphafold/transform.py.
Import your proteins into Elasticsearch.
optional arguments:
-h, --help show this help message and exit
-i I Path to your preprocessed A3M files. THIS ARGUMENT IS MANDATORY.
-n N Elasticsearch index name.
-a Annotate proteins using UniProt API.
-f Force overwrite.
-t T Threads to use.
--max-msa MAX_MSA Max number of msas to use for annotation.
--url URL URL of MineProt Elasticsearch API.
-
If
-nis unset, use basename of-ias-n;Please ensure that your target repository name adheres to the naming standards of Elasticsearch indices.
-
Read each PDB file in the
-idirectory, get their basenames and contents; -
For each PDB file:
-
read related JSON file and generate a JSON request body:
{ "name": PDB basename "score": model score (Usually pLDDT) "seq": protein sequence "anno": { "homolog": homolog accession "database": annotation database (UniProtKB/UniParc) "description": [] } }Please note that your PDB basename will be recognized as protein name.
-
If
-ais set, read related A3M file, enumerate sequence identifiers and send them to the UniProt API, until a UniProtKB/UniParc registered homolog is found:GET https://www.ebi.ac.uk/proteins/api/proteins/<identifier>Set
{anno{homolog}}to the found homolog, and extract its UniProt, GO & InterPro annotations into{anno{description}}; -
Send the request to MineProt Elasticsearch API (
--url):POST <URL>/<N>/add/<base64-encoded_A3M_basename>The URL of Elasticsearch API is usually at /api/es of your MineProt site.
-
Import your proteins into repository.
options:
-h, --help show this help message and exit
-i I Path to your preprocessed files (A3M, PDB, CIF & JSON). THIS ARGUMENT IS MANDATORY.
-n N MineProt repository name.
-m M Upload mode: 0: all files; 1: without A3M; 2: only PDB & JSON; 3: only PDB.
-f Force overwrite.
-z Compress files.
--url URL URL of MineProt import2repo API.
-
If
-nis unset, use basename of-ias-n;Please ensure that your target repository name adheres to the naming standards of Elasticsearch indices.
-
Enumerate each file in the
-idirectory and send their basename to MineProt import2repo/check API:GET <URL>/check.php?repo=<N>&name=<file_basename> -
Skip files already existing in target repository, and import files according to
-mby MineProt import2repo API.The URL of import2repo API is usually at /api/import2repo/ of your MineProt site.
If-fis set, all files will be imported without checking. If-zis set, all files will be gzip-compressed.
In fact, MineProt application can work as usual with only PDB or even FCZ files in repositories. Due to open source licensing issue, MineProt toolkit does not support processing FCZ format. Given the amazing compression power of foldcomp, we recommend you to usersyncto upload FCZ files directly.
These shell scripts integrate their corresponding <AI_system>/transform.py with import2es.py and import2repo.py. Please try <AI_system>/import.sh --help for their arguments.
Export data from MineProt Search Page.
positional arguments:
url Search URL. THIS ARGUMENT IS MANDATORY.
e.g. http://mineprot-demo.bmeonline.cn/search.php?search=dicer
options:
-h, --help show this help message and exit
-o O Path to output folder
- Download
result.jsonfrom MineProt ES Search API hidden in the Search Page to-o; - Read
result.json, make necessary directories in-oaccording to _index (MineProt repositories), and download search result files to corresponding directories.
The -o directory will have the following structure:
<output_dir>/
result.json
<repo1>/
<protein1>.a3m <protein1>.json <protein1>.pdb <protein1>.cif
<protein2>.a3m <protein2>.json <protein2>.pdb <protein2>.cif
<repo2>/
<protein3>.a3m <protein3>.json <protein3>.pdb <protein3>.cif
...
...
Prepare metadata for 3D-Beacons client.
optional arguments:
-h, --help show this help message and exit
-i I Path to your MineProt repo. THIS ARGUMENT IS MANDATORY.
-o O Path to your data directory for 3D-Beacons. THIS ARGUMENT
IS MANDATORY.
-t T Threads to use.
--max-msa MAX_MSA Max number of msas to use for mapping.
-
Make necessary directories in
-o;mkdir -p /path/to/3d-beacons-client/data/{pdb,cif,metadata,index} -
For each PDB file in
-i(usually MineProt repository):-
read related JSON file to get pLDDT;
-
read related A3M file to get mapping UniProt accession, check if it meets the requirement of 3D-Beacons client, pull its sequence and make pairwise alignment;
-
generate JSON in
/path/to/3d-beacons-client/data/metadata:{ "mappingAccession": UniProt accession, "mappingAccessionType": "uniprot", "start": start site of pairwise alignment, "end": end site of pairwise alignment, "modelCategory": "Ab initio", "modelType": "single", "confidenceType": "pLDDT", "confidenceAvgLocalScore": pLDDT, "createdDate": created date of PDB file, "sequenceIdentity": identity of pairwise alignment, "coverage": coverage of pairwise alignment, } -
copy PDB file to
/path/to/3d-beacons-client/data/pdb.
-
You can run
docker-compose exec cli snakemake --cores=2to process your models for 3D-beacons client once this script is done. It's also recommended to directly copy the CIF files in your MineProt repository to/path/to/3d-beacons-client/data/cifand run CLI commands manually.
MineProt plugins are developed to extend the functionality of your protein server.
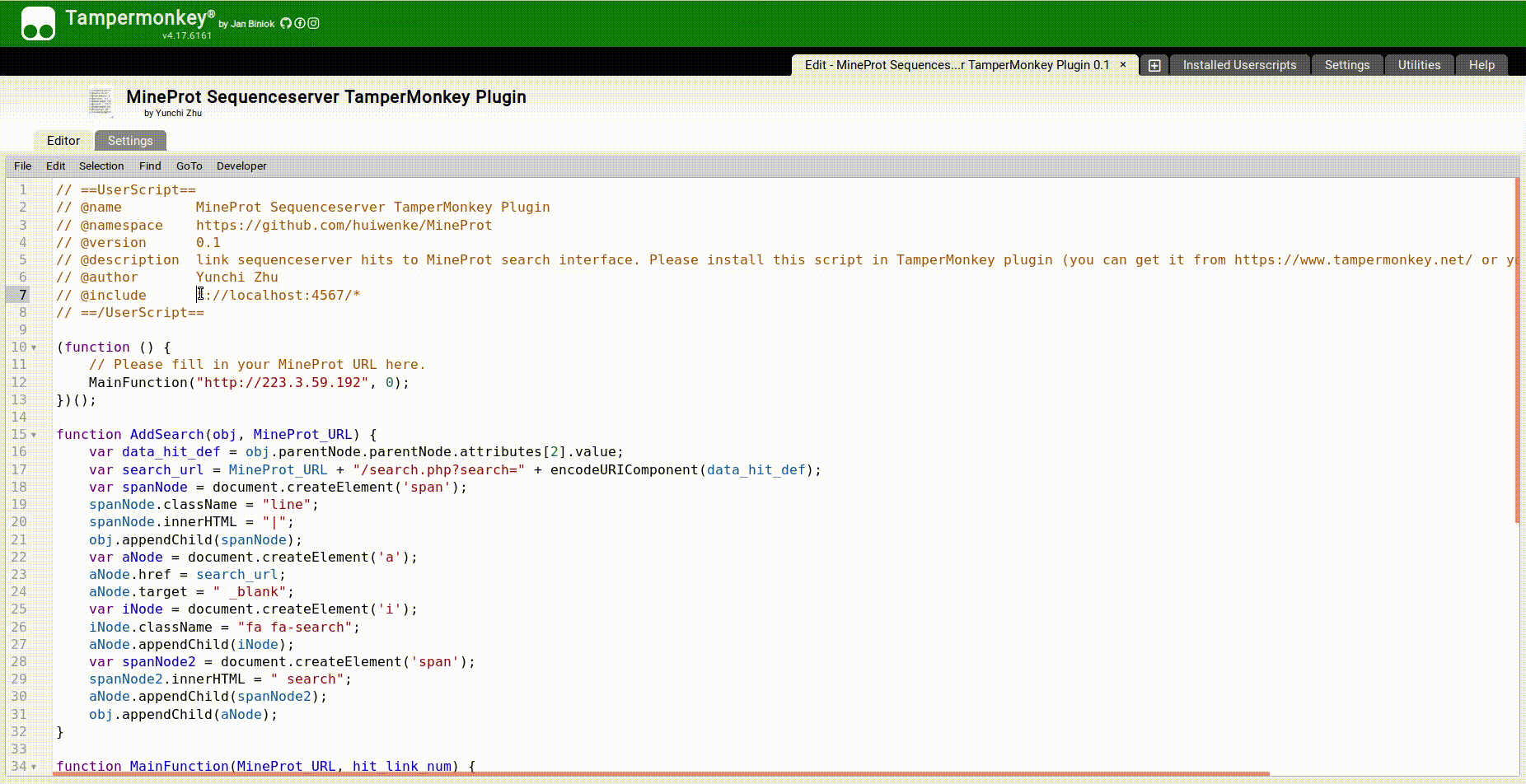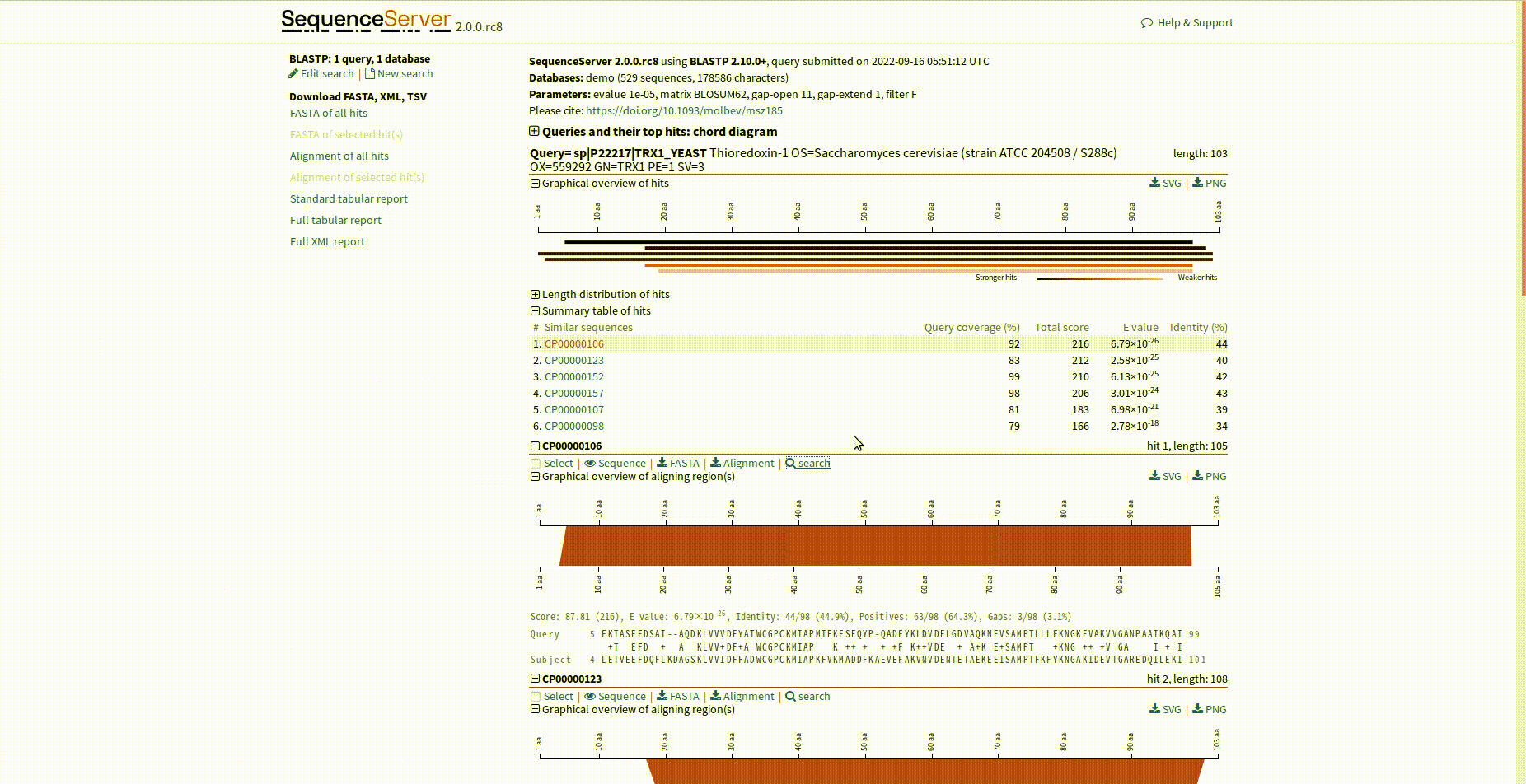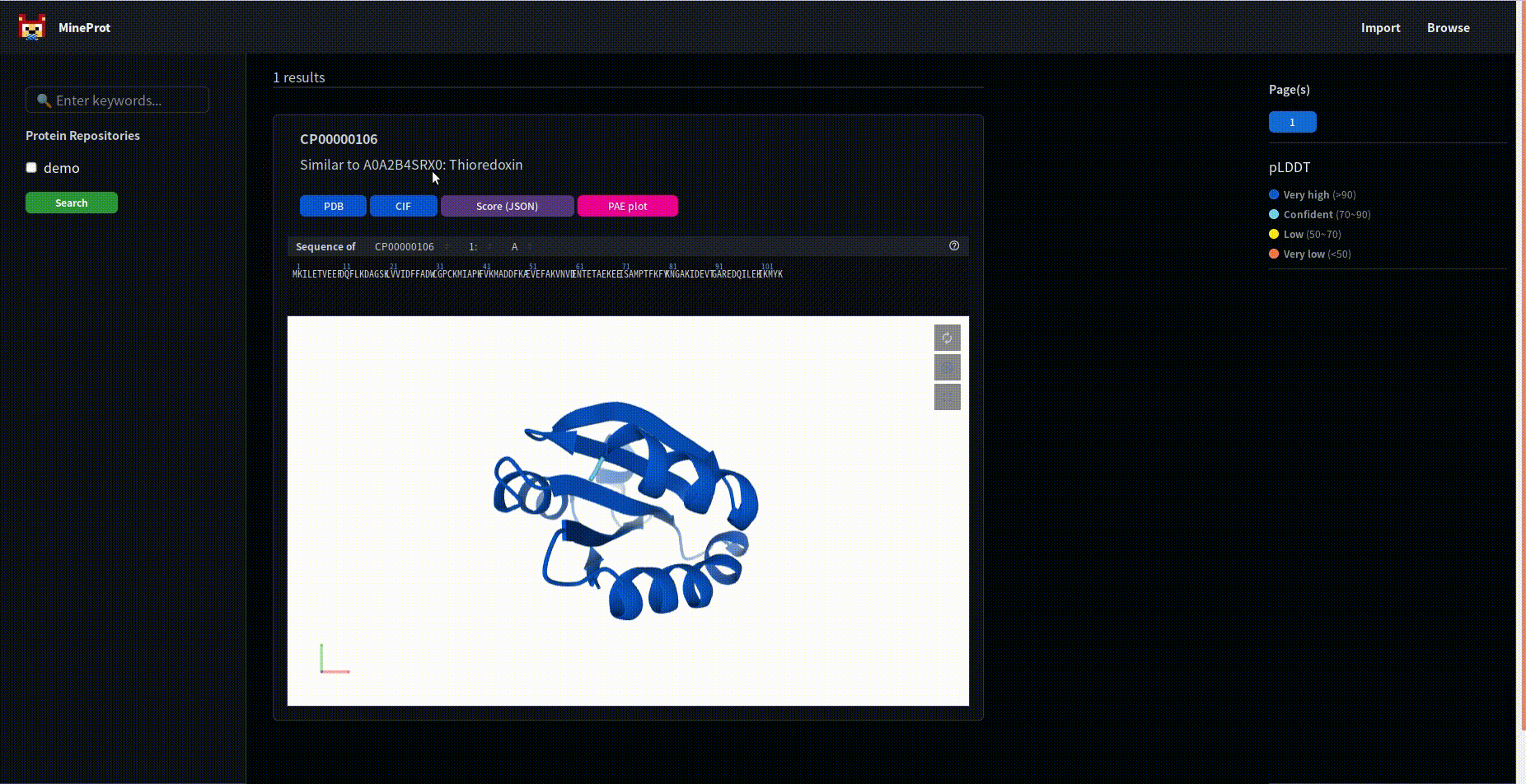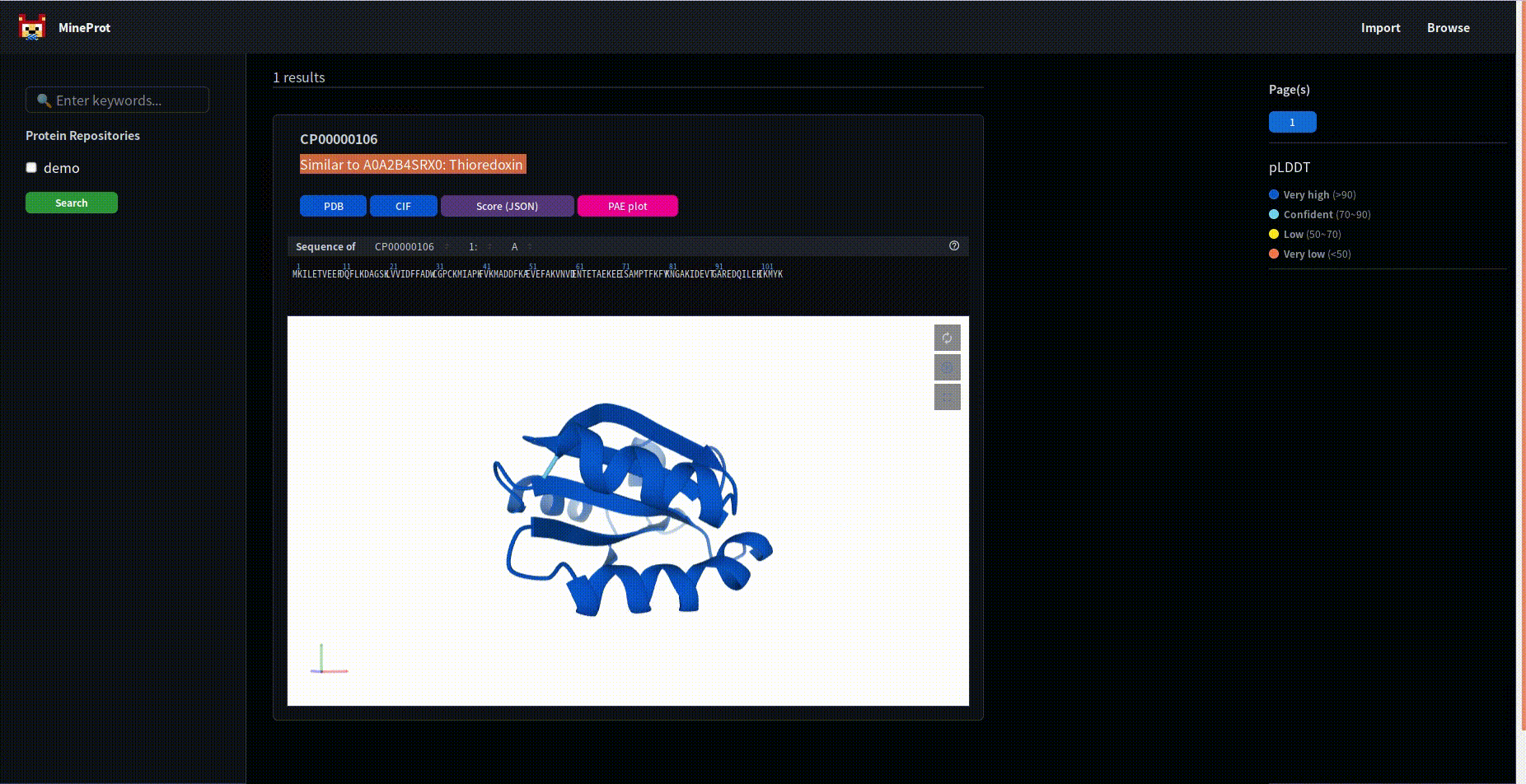
sequenceserver.js is designed to link SequenceServer hits to MineProt search interface, which enables users to search their proteins via BLAST. Please note that the sequence identifiers in your BLAST database should match protein names in your MineProt repositories and be added with suffix "|repo=XXX" for distinction if you have more than one repository. You can use msa2seq.sh to generate FASTA files for SequenceServer deployment.
- Install TamperMonkey plugin to your browser;
- Edit sequenceserver.js:
-
Fill in the URL pattern of target SequenceServer app behind
// @include, for example:// @include *://localhost:4567/* -
Fill in the URL of your MineProt site as the first argument of
MainFunction(), for example:(function () { MainFunction("http://localhost", 0); })();
-
- Add the edited script directly to TamperMonkey.
- Fill in the URL of your MineProt site as the first argument of
MainFunction()in sequenceserver.js; - Add the edited script to views/report.erb of your SequenceServer in a
<script>...</script>tag; - Restart your SequenceServer, and all users can directly access the MineProt search interface of each BLAST hit.
Please note the extra search button.
alphafill.js is designed for directly sending CIF files from MineProt Search Page to AlphaFill online annotation service.
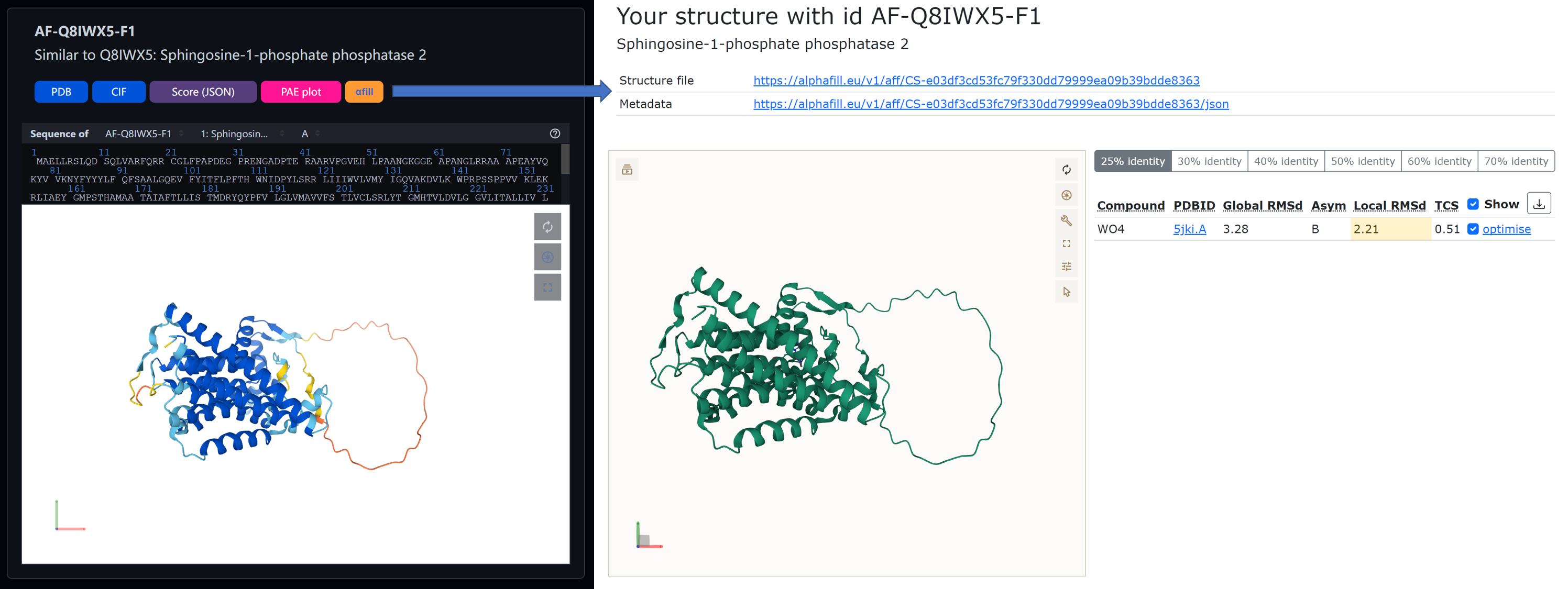
- Install TamperMonkey plugin to your browser;
- Edit alphafill.js:
-
Fill in the URL pattern of your MineProt Search Page behind
// @include, for example:// @include *://localhost/search.php*
-
- Add the edited script directly to TamperMonkey.
Add this script to web/search.php of your MineProt in a <script>...</script> tag.