{kind=link}
##Sewing Machine pipeline: iteratively super scaffold genome FASTA files with BioNano genome maps using stitch.pl

The sewing machine pipeline iteratively super scaffolds genome FASTA files with BioNano genome maps using stitch.pl and the BioNano tool RefAligner until no new super scaffolds can be produced. The pipeline runs alignments with both default and relaxed parameters. These alignments are then used by stitch.pl to superscaffold a fragmented genome FASTA. See tutorial lab to run the sewing machine pipeline with sample data https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/stitch/sewing_machine_LAB.md.
SCRIPT
sewing_machine.pl - a package of scripts that analyze IrysView output (i.e. XMAPs). The script filters XMAPs by confidence and the percent of the maximum potential length of the alignment and generates summary stats of the more stringent alignments. The first settings for confidence and the minimum percent of the full potential length of the alignment should be set to include the range that the researcher decides represent high quality alignments after viewing raw XMAPs. Some alignments have lower than optimal confidence scores because of low label density or short sequence-based scaffold length. The second set of filters should have a user-defined lower minimum confidence score, but a much higher percent of the maximum potential length of the alignment in order to capture these alignments. Resultant XMAPs should be examined in IrysView to see that the alignments agree with what the user would manually select.
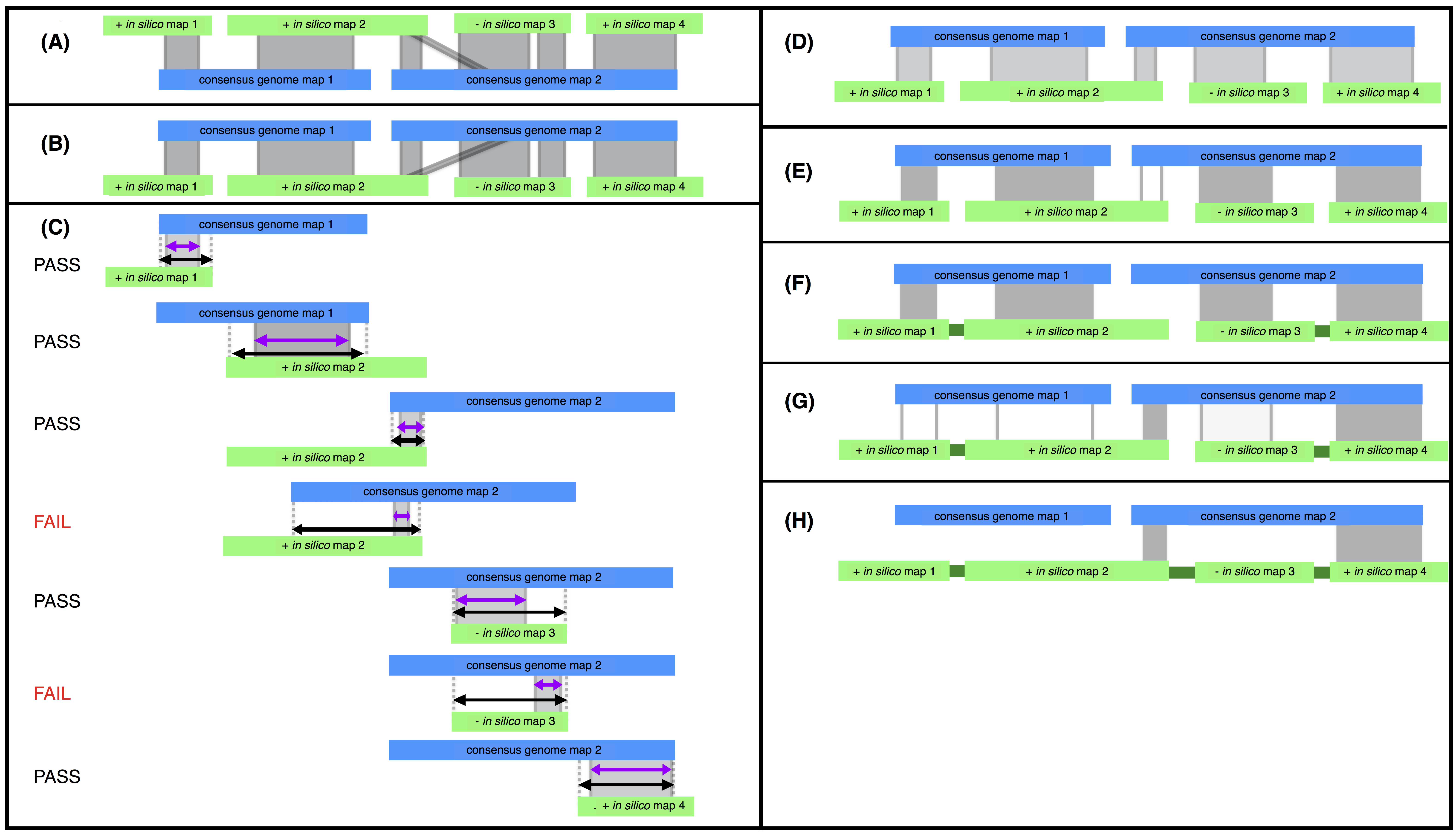
Steps of the stitch.pl algorithm. Consensus genome maps (blue) are shown aligned to in silico maps (green). Alignments are indicated with grey lines. CMAP orientation for in silico maps is indicated with a ”+” or ”-” for positive or negative orientation respectively. (A) The in silico maps are used as the reference. (B) The alignment is inverted and used as input for stitch.pl. (C) The alignments are filtered based on alignment length (purple) relative to total possible alignment length (black) and confidence. Here assuming all alignments have high confidence scores and the minimum percent aligned is 40% two alignments fail for aligning over less than 40% of the potential alignment length for that alignment. (D) Filtering produces an XMAP of high quality alignments with short (local) alignments removed. (E) High quality scaffolding alignments are filtered for longest and highest confidence alignment for each in silico map. The third alignment (unshaded) is filtered because the second alignment is the longest alignment for in silico map 2. (F) Passing alignments are used to super scaffold (captured gaps indicated in dark green). (G) Stitch is iterated and additional super scaffolding alignments are found using second best scaffolding alignments. (H) Iteration takes advantage of cases where in silico maps scaffold consensus genome maps as in silico map 2 does. Stitch is run iteratively until all super scaffolding alignments are found.
DEPENDENCIES
git - see http://git-scm.com/book/ch1-4.html for instructions
BioPerl - see http://www.bioperl.org/wiki/Installing_BioPerl
Requires BNGCompare from https://github.com/i5K-KINBRE-script-share/BNGCompare in your home
directory. Also requires RefAligner. Install BioNano scripts and
executables in `~/scripts` and `~/tools` directories respectively. Follow the Linux installation
instructions in the "2.5.1 IrysSolve server RefAligner and Assembler" section of
http://www.bnxinstall.com/training/docs/IrysViewSoftwareInstallationGuide.pdf to install
RefAligner.
USAGE
Usage: perl sewing_machine.pl [options]
Documentation options:
-help brief help message
-man full documentation
Required options:
-o output directory
-g genome map CMAP file
-p project
-e enzyme
-f scaffold (reference) FASTA
-r reference (in silico map) CMAP file
Required options (for assemble_XeonPhi pipeline):
-b best assembly directory (replaces -o and -g)
Filtering options:
--f_con first minimum confidence score (default = 20)
--f_algn first minimum % of possible alignment (default = 40)
--s_con second minimum confidence score (default = 15)
--s_algn second minimum % of possible alignment (default = 90)
--n minimum negative gap length allowed (default = 20000 bp)
-T RefAligner p-value threshold (default = 1e-8)
Options:
-help Print a brief help message and exits.
-man Prints the more detailed manual page with output details and
examples and exits.
-o, --out_dir
Path of the user selected output directory without trailing
slash (e.g. -o ~/stitch_out ).
-g, --genome_maps
Path of the CMAP file containing genome maps assembled from
single molecule maps (e.g. -g
~/Irys-scaffolding/KSU_bioinfo_lab/sample_output_directory/BioNa
no_consensus_cmap/ESCH_COLI_1_2015_000_STRICT_T_150_REFINEFINAL1
.cmap ).
-p, --project
The project name with no spaces, slashes or characters other
than underscore (e.g. -p Esch_coli_1_2015_000).
-e, --enzyme
A space separated list of the enzymes used to label the
molecules and to in silico nick the sequence-based FASTA file.
They can include BspQI BbvCI BsrDI bseCI (e.g. -e BspQI). If
multiple enzymes were used enclose the list with quotes (e.g. -e
"BspQI BbvCI").
-f, --fasta
Path of the FASTA file that will be super-scaffolded based on
alignment to the assembled genome maps. It is preferable to use
the scaffold FASTA rather than the contigs. Many contigs will
not be long enough to align.
-r, --r_cmap
The reference CMAP produced from your sequence FASTA file.
-b, --best_dir
Path of the user selected directory of the "best" assembly
without trailing slash (e.g.
~/Esch_coli_1_2015_000/default_t_100 ). This parameter replaces
-o and -g when using the assemble_XeonPhi pipeline.
--f_con, --fc
The minimum confidence score for alignments for the first round
of filtering. This should be the most stringent, highest, of the
two scores (default = 20).
--f_algn, --fa
The minimum PAT, or minimum percent of the full potential length
of the alignment allowed, for the first round of filtering. This
should be lower than the setting for the second round of
filtering (default = 40).
--s_con, --sc
The minimum confidence score for alignments for the second round
of filtering. This should be the less stringent, lowest, of the
two scores (default = 15).
--s_algn, --sa
The minimum PAT, or percent of the full potential length of the
alignment allowed, for the second round of filtering. This
should be higher than the setting for the first round of
filtering (default = 90).
-n, --neg_gap
Allows user to adjust minimum negative gap length allowed
(default = 20000 bp).
-t, --p-value_T
The RefAligner p-value threshold (default = 1e-8). Can use -T as
low as 1e-6 for small bacterial genomes or up to 1e-9 or 1e-10
for large genomes (> 1G).
DESCRIPTION
OUTPUT DETAILS:
The script outputs an XMAP with only molecule maps that scaffold in
silico maps and an XMAP of all high quality alignments. Both XMAPs can
be imported and viewed in the IrysView "comparisons" window if the
original r.cmap and q.cmap are in the same folder when you import.
The script also lists summary metrics in a csv file.
In the same csv file, in silico maps that have alignments passing the user-
defined length and confidence thresholds that align over less than 60%
of the total length possible are listed. These may represent mis-
assembled scaffolds.
In the same csv file, high quality but overlaping alignments in a csv
file are listed. These may be candidates for further assembly using the
overlaping contigs and paired end reads.
The script also creates a non-redundant (i.e. no in silico map is used
twice) super-scaffold from a user-provided fasta file and a filtered
XMAP. If two in silico maps overlap on the superscaffold then a 30 "n" gap
is used as a spacer between them. If adjacent in silico map do not overlap
on the super-scaffold than the distance between the begining and end of
each in silico map is reported in the XMAP is used as the gap length. If
a scaffold has two high quality alignments the longest alignment is
selected. If both alignments are equally long the alignment with the
highest confidence is selected.
No in silico map's sequence is added to the final fasta twice; however,
if the first and second best alignment for an in silico map align to the
ends of two molecule maps that each super-scaffold > 1 in silico map than
these alignments are listed and can be used to "stitch" together the final
super-scaffold in a subsequent iteration.
Test with sample datasets
See tutorial lab to run the sewing machine pipeline with sample data https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/stitch/sewing_machine_LAB.md.
UPDATES to stitch.pl
####stitch.pl Version 1.4.7
Fixed bug in halting if no super scaffolds are created and in handling of filenames (to delete intermediate fasta files)
####stitch.pl Version 1.4.6
Automatically skips creating contigs if no super scaffolds were created.
####stitch.pl Version 1.4.5
rejects scaffolding alignments if overlap is longer than the "-n, --neg_gap" argument (default = 20000 bp). This allows user to adjust minimum negative gap length allowed.
####stitch.pl Version 1.4.4
correctly sort new xmaps to correct output order
####stitch.pl Version 1.4.3
changed unknown gap lengths to 100 and set AGP indentity to U from N. Fixed bug in alignments that are not the best or second best.
####stitch.pl Version 1.4.2
fixed bug in splitting sequence at gaps longer than 10 bp into contigs in KSU_bioinfo_lab/stitch/make_contigs_from_fasta.pl
####stitch.pl Version 1.4.1
produces AGP files of super-scaffolds
####stitch.pl Version 1.4.
Speed up KSU_bioinfo_lab/stitch/agp2bed.pl. Modified KSU_bioinfo_lab/stitch/make_contigs_from_fasta.pl to only split at gaps longer than ten bases.
####Update to stitch.pl v.1.3
Die and report when no super scaffolds can be found. Give generic names to contigs. Check for IUPAC ambiguous bases other than n when making AGP.
####stitch.pl Version 1.2
stitch.pl Version 1.2 : Check fasta file for redundant super-scaffold names (this step only effects iterative assemblies), create new AGP, create a BNG compatible Bed file.
####stitch.pl Version 1.1
Separated stitch map from best alignment map. Also generalized agp2bed.pl.